Introduction
Cerebral small vessel disease (SVD) is an important cause of stroke, cognitive impairment, and mood disorder among the elderly.1 It accounts for 15% to 26% of ischemic stroke in the US and Europe.2,3,4,5,6 In Asia, cerebral SVD is responsible for an even greater proportion of ischemic stroke, ranging from 25% to 54%.7,8,9,10,11 In addition, it is the most important cause of intracerebral hemorrhage (ICH)12 and contributes to a substantial proportion of vascular dementia.13
Although traditional risk factors are important in the development of cerebral SVD14 the exact cause is still uncertain because not all individuals with those risk factors develop the disease and often, known risk factors are absent in patients with cerebral SVD.15 Ischemic stroke is possibly a complex genetic disorder in which interactions between candidate genes and environmental factors or established risk factors are important in the development of clinical phenotypes.16 Therefore, genetics may play an important role in understanding the pathophysiology of sporadic cerebral SVD. With advances in genetic methodology such as genome-wide association study (GWAS) and meta-analysis of individual candidate gene study, both, extensive and in-depth information on the susceptible genetic loci associated with sporadic cerebral SVD (Table 1) is now available. In addition, several single-gene disorders causing cerebral SVD have been discovered, including cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), COL4A1-related cerebral SVD, autosomal dominant retinal vasculopathy with cerebral leukodystrophy (RVCL), and Fabry disease (FD; Table 2). Recognition of the genetic aspect of cerebral SVD could lead to improved diagnosis and treatment of these rare single-gene disorders as well as sporadic cerebral SVD. In this review, we have focused on the recent advances in genetics of both, sporadic and familial forms of cerebral SVD.
Genetics of sporadic cerebral SVD
Twin and family history studies
Twin studies have suggested that monozygotic twins were more likely to be concordant for risk of stroke than dizygotic twins and a positive family history was a risk factor for stroke in both case-control and cohort studies.17 For subtypes of ischemic stroke, a family history of stroke was a significant risk factor for large vessel disease (odds ratio [OR], 2.24 to 2.54) and for SVD (OR, 1.93 to 2.62) but not for cardioembolic stroke.18,19 Cerebral white matter hyperintensities (WMH) are one of the main imaging features of SVD, and they are frequently found in patients with lacunar stroke on brain magnetic resonance imaging (MRI).14 Twin studies also indicate a genetic contribution to WMH with concordance rates of 61% in monozygotic pairs and that of 38% in dizygotic pairs for large proportion of WMH.20 The findings were also replicated in the family-based Framingham study, with an overall heritability of approximately 52% to 78%.21 ICH, especially nonlobar ICH, results from the rupture of small penetrating arteries of the brain and is another important clinical manifestation of cerebral SVD.12 Heritability of nonlobar or primary ICH was suggested by case-control studies showing associations between family history and an increased risk of ICH (adjusted OR, 2.1 to 6.3).18,22
Candidate gene studies
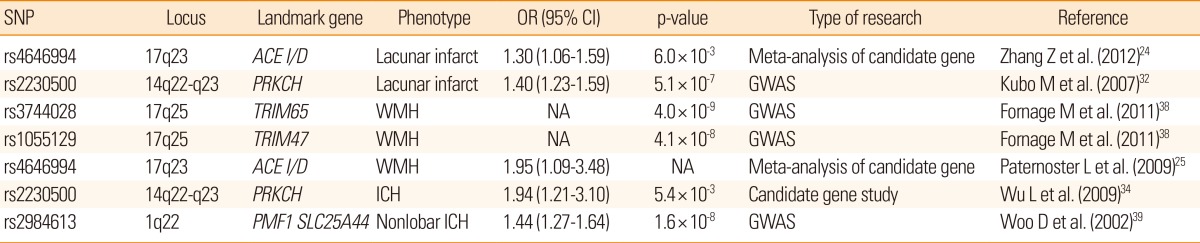
Candidate gene study is an approach involving exploring the genetic influences on a complex trait by identifying candidate genes that might have a role in the etiology of the disease and this method has been widely used for the study of complex diseases.23 In a comprehensive meta-analysis of 187 candidate genetic polymorphism in 37,481 stroke patients, 6 polymorphisms, namely, factor V Leiden, angiotensin converting enzyme (ACE) insertion/deletion (I/D), methylene tetrahydrofolate reductase (MTHFR) C677T, prothrombin G20210A, plasminogen activator inhibitor-1 4G/5G allele, and glycoprotein IIIa L33P, were found to have an association with increased risk of stroke. Among these, ACE I/D polymorphism was more specifically associated with increased risk of lacunar stroke than with other types of ischemic stroke. In another meta-analysis of 50 case-control studies, homozygote DD of ACE gene polymorphism showed a 37% higher risk of ischemic stroke compared with homozygote II and heterozygote ID, and a subgroup analysis showed that the risk was more prominent among Asians and SVD subtype of ischemic stroke.24 Researchers have also looked for associations between polymorphisms in various candidate genes and WMH in brain images. In a meta-analysis including 19 candidate gene polymorphisms from 19,000 subjects, no evidence of association was found between apolipoprotein E (APOE) (ε4+/-), MTHFR C677T, or angiotensinogen M235T, and WMH.25 Only ACE I/D polymorphism showed a significant association (OR, 1.95; 95% CI, 1.09 to 3.48) with the WMH.
Candidate genes that have been investigated most frequently for ICH are APOE, plasminogen activator inhibitor-1 4G/5G, ACE/ID, factor V Leiden, MTHFR C677T, and factor XIII V34L polymorphism.26 In a meta-analyses that included 6,359 cases derived from 55 case-control studies, homozygous for the ACE/I allele and for the 5G allele in plasminogen activator inhibitor-1 4G/5G polymorphism were found to have statistically significant associations with hemorrhagic stroke (including ICH, subarachnoid hemorrhage, vascular malformation, and cerebral aneurysm) when they were analyzed together, but not specifically for ICH. A large-scale genetic association study with 2,189 ICH cases and 4041 controls found that both, the ε2 and the ε4 alleles of APOE were significantly associated with increased risk of lobar ICH. Although, an association was also found between nonlobar ICH and the ε4 allele, the association did not reach predefined significance.27 In a subsequent research, the APOE ε2 allele was also associated with larger ICH volumes, increased mortality, and poorer functional outcomes than non-carriers for patients with lobar ICH.28 In a meta-analyses including 10 studies with 7,351 patients, the APOE ε4 allele was associated with increased risk of cerebral microbleeds in any location compared with common ε3/ε3 genotype (pooled OR 1.22, 95% confidence interval [CI] 1.05 to 1.41, P=0.01), and the association was stronger for strictly lobar, cerebral microbleeds.29 The biological role of APOE genotype in the development of nonlobar ICH or cerebral microbleeds needs further exploration because cerebral amyloid angiopathy-related vasculopathy rarely occurs in vessels of deep gray matter.30
In summary, although meta-analyses of candidate gene studies have overcome an important limitation of small sample size in case of individual case-control studies, these findings should be interpreted with caution, as the results may be confounded by publication bias.
Genome-wide association study
GWAS is a new method to study common genetic variation across the entire human genome, designed to identify genetic associations with observable traits.31 It is particularly useful to investigate common diseases with complex traits such coronary artery disease and stroke. Recently, a genome-wide case-control study revealed that a non-synonymous single nucleotide polymorphism (SNP) (rs2230500) in a member of the protein kinase C family, PRKCH, had a significant association with lacunar infarction in 2 independent Japanese samples (OR, 1.40).32 Later, the same SNP was also found to be associated with silent lacunar infarction.33 This polymorphism increased the risk of both, ischemic and hemorrhagic stroke in the Chinese population.34 Although the study in Chinese population did not specify the subtypes of ischemic stroke, the polymorphism may be specific for cerebral SVD in Asians, because only SVD would explain increased risk of both, ischemic and hemorrhagic stroke in general. The polymorphism may also contribute to a higher proportion of both, lacunar stroke and primary ICH in the Asian population rather than Caucasians, as the minor allele is rarely found in European descendants.32
However, European large-scale collaborative studies applying GWAS technique have confirmed subtype-specific associations for cardioembolic stroke only near PITX2 and ZFHX3 and for large vessel disease at a 9p21 locus and HDAC9, but not for SVD.35 In addition, heritability estimates calculated by genome-wide complex trait analysis were 40.3% for large vessel disease, 32.6% for cardioembolic stroke but only 16.1% for SVD.36 In a meta-analysis of GWAS in Caucasian participants in 6 studies comprising the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium, the most significant association was found on chromosome 20p12 with rs2208454 for silent infarcts found on brain MRI. However, the association did not replicate in independent samples of 1,822 Caucasian and 644 non-Caucasian participants.37 As the candidate genes for cerebral SVD identified in meta-analysis have not been replicated with GWAS, they might have a limited role in lacunar stroke pathogenesis. However, the candidate gene approach can still be useful because the current GWAS cannot cover all alleles with low frequencies.
For cerebral WMH, a meta-analysis of GWAS involving 9,361 stroke-free individuals of European descent from 7 community-based cohorts identified 6 novel SNPs on chromosome 17q25, encompassing 6 known genes. Among these, the significance of two SNPs (rs3744028 and rs1055129) was replicated in an independent sample.38 However, variant alleles at these loci contributed to only a small increase in the WMH burden (4% to 8% of the overall mean WMH burden in the sample).
Recently, a GWAS with 1,545 ICH patients (664 with lobar and 881 with nonlobar) of European descent, reported a significant association between nonlobar ICH and chromosomal region 1q22 (rs2984613, OR=1.44, P=1.6×10-8), which was replicated in another dataset.39 In addition, several other SNPs within this locus also achieved genome-wide significance and some of them were listed in the GWAS report of the WMH mentioned above, which supports a common pathogenesis of both WMH and ICH.38 As described earlier, the 1425G/A SNP (rs2230500) in PRKCH was associated with an increased risk of both, ischemic stroke and hemorrhagic stroke in the Chinese population,34 but the association needs further validation in other ethnic populations.
To summarize, several important genetic loci have been identified for cerebral SVD from various ethnic backgrounds using the GWAS technique. However, detailed information on the specific genes and functional variants should be investigated further to determine the exact role of gene function or gene products in pathogenic mechanisms underlying SVD.
Genetics of single gene disorders causing cerebral SVD
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
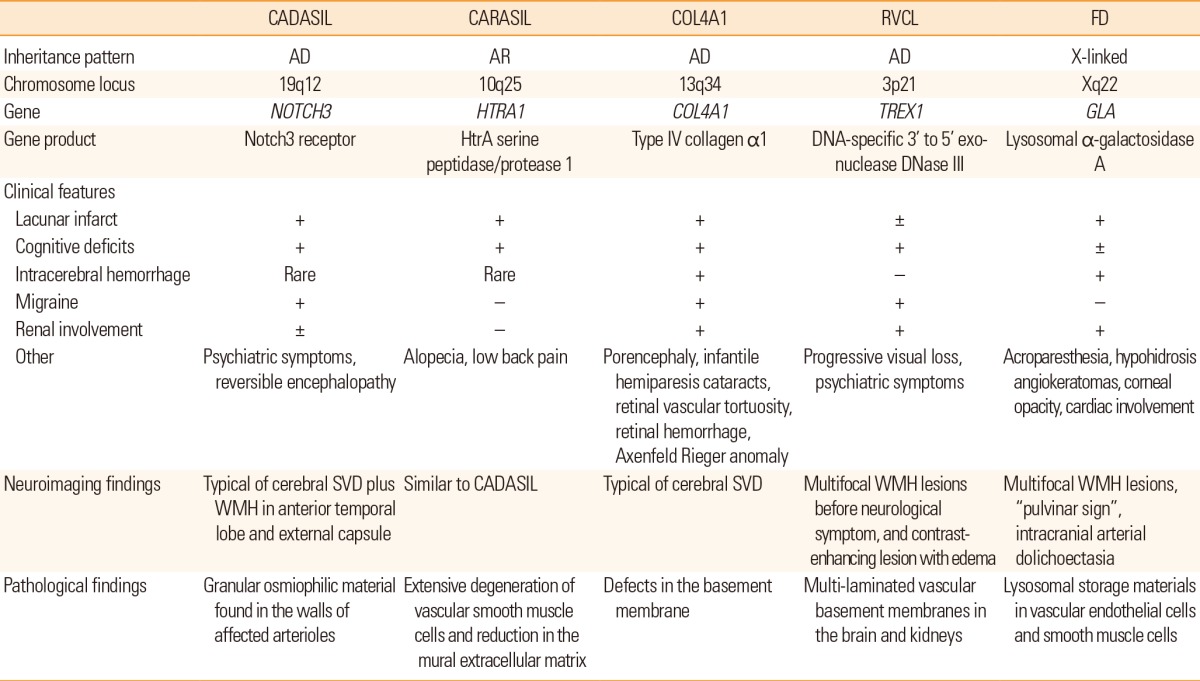
CADASIL is one of the most common single-gene disorders of the cerebral small blood vessels caused by mutations in the NOTCH3 gene on chromosome 19q12.40 The main clinical features include recurrent stroke, migraine, psychiatric symptoms, and progressive cognitive decline.41
Ischemic stroke is the most frequent manifestation and is present in 60% to 84% patients with CADASIL.42,43,44,45 The mean age of stroke onset is around 41 to 49 years and a majority of patients with CADASIL present with typical lacunar syndrome. Cognitive deficit is found in approximately 60% patients, with two-thirds developing dementia by age 65 years. Migraine is the most common initial symptom that occurs in 22% to 77% patients and it usually begins around age 20 years and will develop in 90% patients by age 40 years.42,43,46 Several patients have reported an aura with occasional basilar migraine, hemiplegic migraine, or migraine with prolonged aura. Psychiatric symptoms occur in 20% to 41% patients with CADASIL and mood disorders are most common.44,47 Other infrequent manifestations include epileptic seizures, acute reversible encephalopathy, and neurological complications following catheter angiography.46,48,49 Granular osmiophilic material found in the walls of affected arterioles is a pathologic hallmark of the disease, and it can be observed in skin and muscle, which is useful for pathologic diagnosis.50 Brain MRI of patients with CADASIL frequently shows progressive WMH, multiple lacunar infarcts, and microbleeding.51,52 The involvement of the anterior temporal lobe and external capsule is known to be characteristic in comparison to that in the sporadic form of WMH.53 Recently, a 7-T MRI study found a significant reduction in the number of small veins in the centrum semiovale of CADASIL patients compared with matched controls.54 This significant reduction was detected both within and outside WMH, suggesting that loss of venous vasculature may occur before the development of WMH.
The NOTCH3 gene encodes cell surface receptors that transduce signals between adjacent cells.55
NOTCH3 is mainly expressed in vascular smooth muscle cells in adults and is critical for vascular development, differentiation, and remodeling.56,57 The NOTCH3 receptor is a single-pass transmembrane protein that consists of 2,321 amino acids, and it has a large extracellular domain (ECD), with 34 tandem epidermal growth factor (EGF)-like domains. Each EGF-like domain contains 6 cysteine residues. Currently, over 170 different mutations have been reported in CADASIL and a majority of these are missense, point mutations,58 frequently located in exon 2-24, which encodes the ECD of the NOTCH3 receptor. Frequently, the mutations result in an odd number of cysteine residues added into the affected EGF. However, a small number of studies have reported that the mutations do not involve cysteine. Interestingly, common SNPs that are frequently found at the NOTCH3 gene, apart from the mutations causing CADASIL, also increase the risk of age-related WMH in individuals with hypertension.59
Although CADASIL has been reported worldwide in all ethnic groups, the exact prevalence of CADASIL is unknown. In 2002, the disease prevalence was reported to be 1.98 per 100,000 in West Scotland.60 In a study including 218 subjects with lacunar stroke, CADASIL mutation was found in only 1 patient.61 Recently, our group screened 151 consecutive patients with acute ischemic stroke for NOTCH3 mutations and found that 6 (4.0%, 95% CI, 0.9 to 7.1) had a NOTCH3 gene mutation. The prevalence of CADASIL increased to 36.0% (95% CI, 8.0 to 64.8) for patients with neuroimaging features consistent with advanced SVD.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy
CARASIL is a single-gene disorder of cerebral small blood vessels with autosomal recessive inheritance. This disorder is caused by mutations in the HTRA1 gene encoding HtrA serine peptidase/protease 1 (HTRA1).62,63 Since the first report of encephalopathy without hypertension from a Japanese family in 1976, the disorder has been predominantly reported in Japan.64 A small number of cases in patients from other ethnicities have also been reported recently.65,66 Although the exact prevalence is unknown, CARASIL is much rarer than CADASIL, as approximately only 50 patients have been reported so far.63
The main clinical features include early-onset lacunar stroke, progressive cognitive deficit, gait disturbance, alopecia, and low back pain.63,64 Lacunar stroke, reported in approximately 50% patients, is the most common manifestation of CARASIL and is usually found in the basal ganglia or brainstem. Cognitive deficit is the second-most common symptom and almost all patients develop dementia by 30 to 40 years of age, which is significantly earlier than CADASIL. Alopecia is the most frequent initial manifestation of the disorder, which can be found in almost 90% patients. It usually begins at adolescence and involves diffuse alopecia involving the entire scalp. Low back pain develops in approximately 80% patients and usually begins at 20 to 40 years of age. Radiologically, spondylosis deformans or disk degeneration has been identified at the cervical or thoracolumbar junction. Unlike CADASIL, migraine-like headache has not been reported with CARASIL; however, cranial MRI findings are reported to be similar to that in CADASIL patients. Pathologically, extensive degeneration of vascular smooth muscle cells and reduction in the mural extracellular matrix are found in cerebral small arteries; however, granular osmiophilic materials or amyloid deposition has not been identified.
HTRA1 is a serine protease that inhibits signaling by members of the transforming growth factor β (TGF-β) family.62 To date, 10 mutations in the HTRA1 gene have been identified in 12 families, resulting in decreased level of protease activity leading to an increase in TGF-β signaling.64 Abnormally increased TGF-β signaling seems to cause degeneration of vascular smooth muscle cells, because TGF-β plays an important role in the differentiation of vascular smooth muscle cells.
COL4A1-related cerebral SVD
Mutations in a gene encoding type IV collagen α1 (COL4A1) were initially associated with porencephaly and infantile hemiparesis,67 however, these mutations have been later found to cause cerebral SVD even in adulthood.68,69 In a systemic review of 52 carriers of the COL4A1 mutation, stroke was present in 9 (17%) subjects in addition to infantile hemiparesis; 6 cases showed ICH and 3 cases had lacunar stroke.70 The mean age of onset of stroke was 36 years (range, 14 to 49 years), and ICH was often recurrent and provoked by minor trauma, activity, or anticoagulant use. Asymptomatic intracranial aneurysm was also found frequently, while migraine was reported in 30% cases with a mean age of onset at 30 years. In addition, several cases of systemic involvement were also found in the eye (cataracts, retinal vascular tortuosity, retinal hemorrhage, Axenfeld-Rieger anomaly), kidney (hematuria, renal cysts), and muscles (cramps). Type IV collagen is the main component of all basement membranes in humans and accordingly, mutations in COL4A1 contribute to a broad spectrum of disorders involving the brain, muscles, eyes, and kidneys. The COL4A1 gene consists of 52 exons and is located in chromosome 13q34.71 More than 30 types of mutations have been reported to date in the COL4A1 gene, and the number is rapidly increasing.72 Although COL4A1 gene mutations were known to cause instability and defects in the basement membrane, the exact mechanism linking mutations and phenotypes is presently unknown.
Retinal vasculopathy with cerebral leukodystrophy
Hereditary vascular retinopathy, cerebroretinal vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS) were originally reported as distinct autosomal dominant disorders.73,74,75 In addition to vascular retinopathy as a common feature, the disorders present with various combinations of Raynaud phenomenon, migraine, cranial pseudotumor, and mild kidney or liver dysfunction. Subsequently, three disorders were mapped to a common locus in chromosome 3p21, and these are now collectively termed as autosomal dominant RVCL.76 The disorder is caused by C-terminal heterozygous, frame-shift mutations in TREX1 gene encoding a DNA-specific 3' to 5' exonuclease DNase III.77 Interestingly, homozygous mutations within this gene cause Aicardi-Goutieres syndrome, which presents as severe encephalopathy, calcification of the basal ganglia, and leukodystrophy.78 Currently, the exact mechanism underlying the mutations in TREX1 causing clinical manifestation is uncertain. The TREX1 proteins found in individuals with RVCL lack part of the C-terminus and this finding might be associated with its intracellular localization.
In a Chinese family with 11 affected patients with HERNS, progressive visual loss began at 20 to 30 years of age and other symptoms included psychiatric symptoms, migraine, and renal dysfunction.74 Neurologic deficits usually occurred as stroke-like episodes following visual loss and consisted of multifocal cortical and subcortical dysfunction. Brain MRI showed multifocal WMH lesions before neurological symptoms, which later turned into contrast-enhancing lesions with edema, frequently found at the time of focal neurologic deficit. Electron microscopic examination revealed distinctive multi-laminated vascular basement membranes in the brain and kidney, and fluorescein retinal angiography frequently showed juxtafoveolar capillary obliteration with tortuous telangiectatic microaneurysms.
Fabry disease
FD is an X-linked, inherited disorder affecting glycosphingolipid metabolism, caused by absent or deficient lysosomal α-galactosidase A activity.79 This enzyme deficiency results in accumulation of globotriaosylceramide within the lysosomes of various organs, such as blood vessels, kidneys, heart, and dorsal root ganglia. Lysosomal α-galactosidase A is encoded by the GLA gene on chromosome Xq22, and more than 585 pathogenic mutations have been reported in the GLA gene to date. FD incidence has been estimated to be 1 per 17,000 to 117,000 in the general population,80,81 but the prevalence ranges from 0.6% to 11.1%, with an average of 4.5% in men and 3.4% in women among patients with cryptogenic stroke.82
The classic form of FD, with no detectable α-galactosidase A presents initially with angiokeratomas, acroparesthesia, hypohidrosis, corneal opacity in childhood or adolescence, and subsequently, progressive vascular disease of the heart, kidneys and brain. Burning pain that typically occurs in the extremities (acroparesthesia) is present in 60% to 80% patients, and autonomic dysfunctions such as hypohidrosis, cardiac arrhythmia, or intestinal motility disorder occurs in patients with autonomic neuropathy. Cerebrovascular complications usually develop in the fourth decade and a majority of patients with FD present with posterior circulation stroke unlike the frequency of ischemic stroke in the general population.83 In addition to SVD, patients with FD may exhibit various stroke mechanisms such as cardiogenic embolism due to ischemic heart disease or valvular heart disease, and prothrombotic state. Recurrent strokes are commonly reported, occurring in 76% to 86% patients. Elongated, ectatic, tortuous, vertebral and basilar arteries are the most common angiographic findings. Almost all patients show WMH on brain MRI at more than 55 years of age84 and the pattern of WMH does not differ significantly from that of sporadic cerebral SVD. Other evidences of cerebral SVD including lacunar infarcts, and microbleeds have also been demonstrated in patients with FD, with the so-called "pulvinar sign" on MRI, namely hyperintensity in the pulvinar on T1-weighted images, reported in 23% patients with FD.85
Clinical and therapeutic applications
Since the introduction of the GWAS technique, more accurate models for risk prediction of complex disorders, based on genetic profiles are expected.86 However, genetic risk scores predicting future stroke based on genetic variants associated with stroke and its risk factors have been discouraging to date, as the models provide only a small improvement in risk prediction compared with the classical epidemiological risk factors for stroke.87,88
For rare, single-gene disorders causing cerebral SVD, timely diagnosis is important for patients and treating physicians because such patients usually have recurrent strokes that lead to worse outcomes at a young age than patients with sporadic cerebral SVD. Genetic counseling is crucial for patients and family members as part of psychological support, reproductive planning, and education of genetic testing. As effective treatments are unavailable for most of those patients at present, routine screening of asymptomatic family members is not recommended. For patients with CADASIL, conventional cerebral angiography has been reported to cause neurological complications in up to 70% patients, and therefore, other forms of angiography such as CT or magnetic resonance angiography should be considered first in patients with CADASIL.48
Because the occurrence of ICH during birth decreased dramatically with surgical delivery in an animal model for COL4A1 mutation,68 a cesarean delivery is recommended for pregnant women carrying the COL4A1 mutation to reduce birth trauma. Patients with COL4A1 mutation should also avoid other known risk factors for ICH such as head trauma or use of anticoagulants. At present, the available data suggest that enzyme replacement therapy, either agalsidase α or β, is possibly effective only for reducing neuropathic pain but not for preventing cerebrovascular complications in patients with FD.89
Conclusion and future perspective
Although several epidemiological studies suggest the heritability of sporadic cerebral SVD, candidate gene studies have been disappointing in identifying specific genes causing the disorder. Several important genetic loci have been identified for cerebral SVD by using the GWAS technique; nevertheless, second-generation GWAS would need an even larger sample size (e.g., 100,000 to 200,000 for a complex disease such as stroke), because undiscovered susceptibility loci are expected to have smaller effect size than the loci that are known.90 To obtain such a large sample size, stroke researchers from various regions of the world have set up the International Stroke Genetics Consortium (ISGC, www.strokegenetics.org). Recently, the consortium published recommendations for phenotypic data collection, biological sample collection, and storage to maintain standardization, reliability, and quality of the data.91,92 More active participation in the international collaborative network is required from Asian countries considering the high incidence and prevalence of stroke in this region.
With the development of next-generation genotyping and sequencing platforms, DNA sequencing costs have declined dramatically in recent years,93 which would facilitate the detection of rare single-gene disorders causing cerebral SVD. In addition, the exact role of common variants within genes causing rare cerebral SVD needs further investigation, because common variants of the NOTCH3 gene show an increased risk of age-related WMH lesions among the elderly. Rare single-gene disorders can help understand the pathogenesis of the more common, sporadic form of cerebral SVD by identification of cellular, molecular, and biochemical changes underlying cerebral small vessel damage through animal models. Improved mouse models that can express the full spectrum of human cerebral SVD need to be developed to help develop targeted treatment strategies based on the pathomechanism underlying vascular damage in cerebral SVD. For example, the clearance of NOTCH3 aggregates or the reduction of mutant NOTCH3 expression could be considered as treatment for patients with CADASIL in the near future.94