Introduction
Cerebral small vessel disease (CSVD) is a series of clinical, imaging, and pathological changes caused by various cerebrovascular diseases. The most common pathological changes in CSVD include arteriosclerosis and cerebral amyloid angiopathy (CAA). In recent years, with improvements in imaging technologies and the discovery of new imaging markers, the diagnostic rate of CAA has significantly improved. CAA is a cerebrovascular disease characterized by the deposition of amyloid β (Aβ) in the vascular walls of the intracranial microvessels. Neuropathological examination has shown that Aβ is mainly deposited in the cerebral and cerebellar meningeal arterial walls and cerebral cortical arteriolar walls. Other regions, such as the cerebellar cortex, thalamus, and brainstem, may also be involved [1].
CAA is an important risk factor for spontaneous intracerebral hemorrhage (ICH) and is closely associated with age-related cognitive impairment [2]. To date, the pathogenesis of CAA remains unclear despite extensive in vivo and in vitro research, and related treatment methods are still in the exploratory stage. Based on the latest research worldwide, this review describes the etiology, epidemiology, pathophysiological mechanisms, clinical characteristics, imaging manifestations, imaging markers, diagnostic criteria, and treatment of CAA to provide a new direction for the clinical prevention and treatment of CAA.
Etiology and epidemiology of CAA
CAA is very common in the older population and its prevalence increases with age. A longitudinal clinical and pathological study on aging indicated CAA-related pathological changes in nearly 80% of older individuals (the average age of death was 88.5±6.6 years) [3]. Autopsy studies on elderly people in the community have confirmed that about one-third of the elderly have the same neuropathological changes as CAA, and moderate to severe pathological changes are considered to be closely related to cognitive changes in patients [4]. These studies show that CAA is closely associated with age and AD.
CAA comprises three main types: hereditary, sporadic, and iatrogenic (also known as acquired); among these sporadic CAA (sCAA) is the most common, whereas iatrogenic is a newly discovered type [5,6]. Hereditary CAA mainly includes Dutch-type hereditary cerebral hemorrhage with amyloidosis (HCHWA-D), Icelandic patients with hereditary cerebral hemorrhage with amyloidosis (HCHWA-I), and Italian-type CAA [7]. HCHWA-D is an autosomal dominant genetic disease caused by a point mutation in the β amyloid precursor protein (APP) gene on chromosome 21 [7]. This mutation leads to amino acid substitution at codon 693 (E22Q), which leads to changes in Aβ cleavage and secretion, enhanced aggregation characteristics, enhanced proteolysis resistance, decreased affinity of brain efflux transporters, and enhanced cell surface binding, ultimately affecting cell homeostasis [8,9]. HCHWA-I is caused by point mutations in cystatin C [10,11].
Sex may also play a key role in CAA progression. A study of patients with HCHWA-D and sCAA found that male sex and sCAA may be related to an earlier onset and longer course of CAA [12].
Pathophysiological mechanisms underlying CAA
Imbalance between Aβ production and elimination
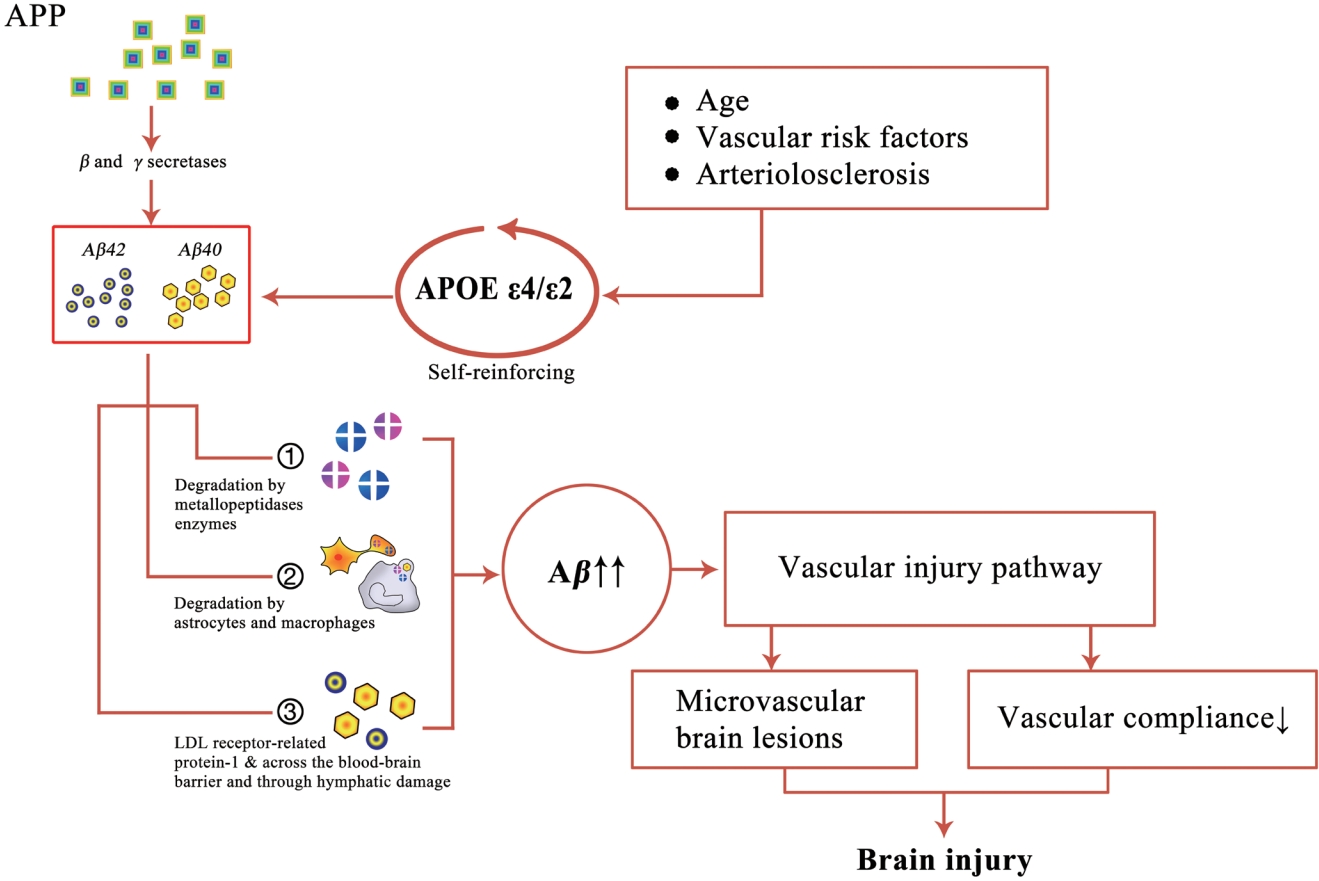
Aβ is a peptide composed of 36-43 amino acids, which is produced by β and γ secretase-mediated APP cleavage. Aβ plays an important role in the occurrence and development of CAA and AD, both of which are associated with similar pathogenic pathways, namely Aβ production, metabolism, perivascular drainage and clearance, and deposition in the periarterial drainage pathway. However, there are many differences between the two. Regarding Aβ deposition, Aβ in CAA is deposited in the cortex and pia mater (mainly arteries, arterioles, capillaries, and veins) vascular walls, whereas in AD, Aβ is mainly deposited in the brain parenchyma [13]. At the molecular level, Aβ40 is mainly associated with CAA, whereas Aβ42 is associated with AD [14,15]. According to the deposition site of Aβ, CAA can be categorized as CAA type 1 (also known as capillary CAA), which is associated with the apolipoprotein E (APOE) ε4 allele, and CAA type 2, which is associated with the APOE ε2 allele [16]. In CAA type 1, Aβ is mainly deposited in cortical capillaries, whereas in CAA type 2, Aβ is commonly found in the pia mater and cortical arteries, arterioles, and veins [7]. Aβ deposition in blood vessels is mainly divided into three stages: in the first stage, Aβ is mainly deposited in cortical and pia mater vessels in the neocortical area; in the second stage, Aβ deposition spreads to the cortex and cerebellum; and in the third stage, Aβ deposition spreads to the basal ganglia, diencephalon, brain stem, and/or white matter [17]. Under normal physiological conditions, soluble Aβ is removed by various clearance systems. However, this balance is compromised in some pathological conditions, leading to abnormal deposition of Aβ in the basement membrane of some small vessels, thus promoting the occurrence and progression of small vessel diseases such as CAA.
In addition, studies have shown that perivascular spaces (PVSs), mainly located in perforating cortical arterioles, are closely related to CAA severity [18]. Moreover, the destruction of vascular structure and function resulting from Aβ deposition slows down the clearance rate of Aβ, forming a vicious circle of clearing obstacles (Figure 1).
APOE mutation
Apolipoproteins in the brain tissue are mainly produced in neurons, astrocytes, and microglia, which can bind to Aβ and other plasma proteins and lipids and be excreted through PVSs and the basement membrane. APOE is a long-chain protein composed of 299 amino acids that is mainly synthesized in the liver, brain, and kidneys [19]. It transports lipoproteins, liposoluble vitamins, and cholesterol to the lymphatic system and blood. APOE primarily consists of three alleles: ε2, ε3, and ε4, which encode the three main subtypes of APOE proteins: APOE2, APOE3, and APOE4, respectively [20]. Compared with ε2 and ε3, APOE ε4 promotes the diffusion and aggregation of Aβ into oligomers and fibrils, affects Aβ clearance, and enhances amyloid plaque formation and deposition with Aβ as the core, ultimately affecting CAA occurrence and progression [21]. Large-scale clinical evidence has shown a close relationship between APOE status and CAA-related ICH. A meta-analysis evidence showed a dose-dependent association of APOE ε4 with sCAA occurrence [22].
APOE has also been identified as a major genetic risk factor for cognitive decline and delayed AD. Most studies have shown that APOE ε4 is associated with pathology including AD and CAA. Compared with ε4 carriers, the incidence of CAA in ε2 carriers is significantly higher, while AD-related pathological changes are fewer [23].
CAA-related inflammation
CAA-related inflammation (CAA-ri) is a rare type of CAA, which refers to an inflammatory reaction of mononuclear macrophages or lymphocytes in or around the walls of Aβ-deposited cortex and leptomeningeal vessels, is accompanied by multiple microinfarcts and hemosiderin deposition and is also known as Aβ-associated vasculitis. The pathological subtypes of CAA-ri include inflammatory CAA (ICAA) and Aβ-related angiitis (ABRA) [24,25]. Presently, the specific pathogenesis of CAA-ri is unclear, which may be related to the autoimmune inflammatory reaction caused by the abnormal deposition of Aβ amyloid protein.
The clinical manifestations of CAA-ri mainly include subacute neurobehavioral symptoms, headaches, seizures, and stroke-like signs, in sharp contrast to the acute onset of ICH commonly observed in CAA [26]. Focal neurological dysfunction was the most common symptom of CAA-ri (76%), followed by cognitive decline (46%) and headache (34%) [27]. But unlike ICAA, more than half of the patients with ABRA showed changes in mental state (59%), 35% had headache as the primary symptom, and 12% had hallucinations [28]. The risk factors for CAA-ri may include age and the APOE ε4/ε4 genotype, and the latter is closely associated with vasculitis or perivascular inflammation [26,29]. Renard et al. [30] found that approximately 71%-77% of patients with CAA-ri carried the APOE ε4/ε4 gene in contrast to 4%-5% of patients with CAA.
A recent study showed that, compared to other non-inflammatory CAA, CAA-ri has a higher amyloid load and more cerebral microbleeds (CMBs). Untreated CAA-ri can lead to more serious disease, worse clinical outcomes, and more severe loss of autonomic function [26]. Different from simple CAA and other diseases that need differential diagnosis, including central nervous system infection and tumor, CAA-ri responds favorably to immunosuppressive therapy; hence, early diagnosis and active treatment are particularly important. Corticosteroids or immunosuppressants are mainly used to treat CAA-ri and can significantly improve the clinical symptoms of some patients.
Other factors
Some studies have shown that other factors play important roles in the occurrence and development of CAA. High levels of β-site APP-cleaving enzyme 1 in endothelial cells can damage the integrity of tight junctions and cause endothelial dysfunction, which further leads to vascular hypoperfusion, blood-brain barrier leakage, microhemorrhage, microvascular degeneration, and neuronal dysfunction, thus aggravating the development and evolution of CAA [31]. In addition, endothelial nitric oxide synthase (eNOS) has been confirmed to play a key role in accelerating the clearance of Aβ and inhibiting its production, thus reducing the deposition of Aβ in cerebral microvessel walls, while simultaneously promoting vasodilation and preventing the increase in cerebral blood pressure, thus reducing cerebral microhemorrhage [31-33]. A compromise in this balance leads to Aβ deposition, elevated blood pressure, and cerebral microhemorrhage, further promoting CAA and AD progression. Moreover, some risk factors related to AD pathogenesis have been identified in patients with CAA, including presenilin, enkephalinase, and zinc ions, and their roles in the pathogenesis of CAA are also being studied.
Clinical characteristics of CAA
The main clinical manifestations of CAA include spontaneous lobar hemorrhage, cognitive impairment, and dementia (after cerebral hemorrhage or in non-stroke patients), as well as transient focal neurological episodes (TFNEs), often associated with acute convex subarachnoid hemorrhage (cSAH) or superficial cortical iron deposition [34].
Intracerebral hemorrhage
ICH is mainly caused by the bleeding of small arteries into the brain parenchyma, caused by CSVD, and is the most common manifestation of CAA. The most common sporadic small vessel diseases that cause ICH are deep perforating artery disease (also known as hypertensive arteriopathy or arteriosclerosis) and CAA. Deep perforating artery disease is mainly related to hypertension, which is a common cause of deep cerebral hemorrhage in the basal ganglia or brainstem, whereas CAA is caused by Aβ deposition in cortical and leptomeningeal vessels, which leads to lobar hemorrhage [35].
The average age at CAA-ICH onset is 73.2 years, and the number of cases increases with age [36]. Its clinical manifestations often vary depending on the size, location, and extent of bleeding, as well as the severity of the previous brain injury. Similar to hypertensive ICH, the clinical manifestations of CAA-ICH mainly include focal neurological manifestations, such as seizures, transient hemiplegia, consciousness disorders, aphasia, and visual field defects [37]. Lobar hematoma is the most serious complication of CAA, with a total mortality rate of 10%-40% in patients with CAA-related ICH and 10% risk of recurrence annually [38].
Some studies have shown that hypertension is not the most important risk factor for CAA-ICH, but that the presence of hypertension can increase the incidence of intracerebral hematoma in patients with CAA [39]. Using new imaging technologies to correctly evaluate will greatly improve disease diagnosis and treatment.
Transient focal neurological episodes
TFNEs, also known as amyloid seizures, are mostly recurrent, stereotyped, and transient (mostly no more than 10-30 min) and are accompanied by diffuse facial and hand sensory abnormalities [40,41].
The main positive TFNE symptoms are “premonitory” diffuse sensory abnormalities/positive visual phenomena or limb convulsions, whereas the main negative symptoms are “transient cerebral ischemia paroxysmal” sudden limb weakness, dysphasia, or vision loss [41]. Since both TFNE and transient ischemic attacks (TIA) manifest similar symptoms of neurological deficits, it is difficult to distinguish between them. However, TIA is usually characterized by transient negative symptoms, such as aphasia, fatigue, and hypoesthesia, whereas TFNE is mainly characterized by positive symptoms [42], which are mainly related to cortical spreading depolarization (also known as diffusion inhibition), corresponding to superficial hemorrhagic lesions associated with CAA [42]. In addition, the pathogeneses of the two are quite different; the former is a cerebrovascular ischemic event, whereas the latter involves leptomeningeal and cortical small blood vessel rupture and bleeding. The induction of CAA-related TFNE may be due to superficial cortical hemorrhage, which is called cSAH in acute cases and cortical superficial siderosis (cSS) in chronic cases. Currently, cSS is considered the strongest clinical marker of TFNE [43,44].
Antiplatelet therapy (APT) and anticoagulant therapy (ACT) may increase the risk of ICH, which may be caused by both CAA-related TFNE and cSS [45]. Therefore, it is necessary for CAA patients with TFNE to discontinue antiplatelet drugs to reduce the risk of lobar hemorrhage. Currently, research on CAA-related TFNE is mainly focused on improving the accuracy of diagnosis and treatment; thus, identifying suitable biological markers may improve this situation.
Cognitive impairment and dementia
Both CAA and AD exhibit abnormal amyloid deposition at the pathological level, and their comorbidity is very common in clinical practice. Autopsy evidence suggests that 80%-100% of patients with AD have deposition of CAA-like amyloid in their intracranial blood vessels, while 40%-60% of patients with CAA-ICH have pathological changes similar to those in AD in their brains [46].
Presently, the mechanism underlying the frequency and severity of cognitive impairment/dementia in patients with CAA is still unclear. Most symptoms of mild CAA cognitive impairment are not obvious. Cognitive impairment in moderate and severe CAA is mainly characterized by impairment of perceptual speed and episodic memory, rather than semantic memory, working memory, and visual spatial dysfunction [47]. Some scholars believe that reducing white matter damage, increasing cerebrovascular reactivity, and preventing cSS occurrence may have a positive regulatory effect on reducing CAA-induced cognitive impairment [48]. In addition, Rabin et al. [49] have shown that the association between severe CAA and a higher Aβ load can indirectly promote cognitive decline by increasing the deposition of tau protein. These findings suggest that tau protein can also drive cognitive impairment through AD-related pathways.
CAA imaging manifestations and imaging markers
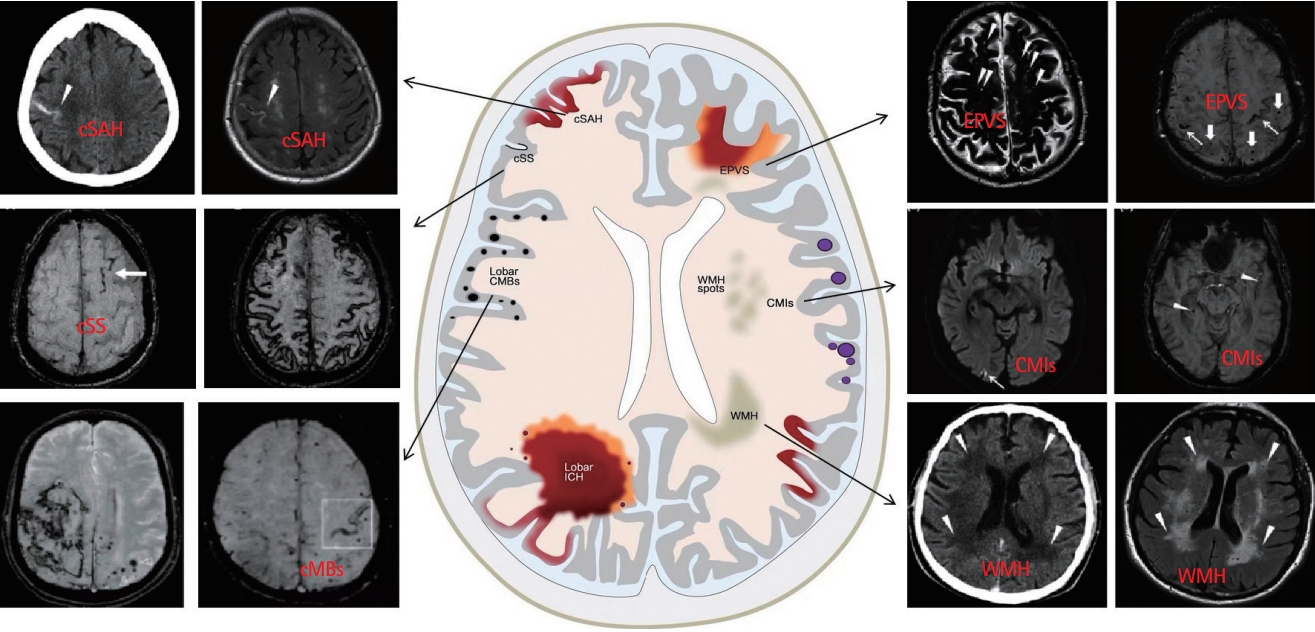
Imaging findings of CAA mainly include two categories: bleeding signs, such as ICH, CMBs, and cSS, and non-bleeding signs, including white matter hyperintensity (WMH), enlarged PVS (EPVS), and cerebral microinfarction (CMI) (Figure 2) [50,51]. Some scholars believe that cortical atrophy [52] and lacunar infarction [53] are characteristic imaging findings of CAA; however, these findings remain controversial. Further in-depth research is required to explore their roles in CAA.
Cerebral microbleeds
CMBs are focal deposits of blood degradation products (including hemosiderin granules) in macrophages, which are caused by the decomposition of hemoglobin released from red blood cells after the degeneration of small vessels, resulting in blood extravasation. CMBs mainly occur in areas with a rich distribution of small blood vessels, such as the basal ganglia, thalamus, and cerebellum [54], which can affect the normal physiological function of brain tissue and lead to neurological functional decline. In general, CMBs have single or multiple, circular or quasi-circular, homogeneous, and well-defined signal missing areas with diameters of 2-5 mm (usually <10 mm) on susceptibility-weighted imaging and gradient recalled echo (GRE) T2*-weighted imaging [53,55].
CMBs are most common in the temporal-parietal cortex and preferentially occur in areas where amyloid deposits are locally increased, resulting in decreased executive function, slower information processing, and lower Mini-Mental State Examination scores [56]. In addition, the results of The Rotterdam Scan Study indicate that APOE ε4 alleles are closely related to the occurrence of multiple CMBs, and age is positively correlated with CMB occurrence [57].
White matter hyperintensity
WMH is an important imaging sign of CSVD that shows high-signal intensity on T2 and fluid-attenuated inversion recovery (FLAIR) sequences [58]. WMH includes two main categories: deep subcortical and periventricular WMH [59]. The severity of white matter lesions is typically evaluated using the Fazekas grading scale.
Xu et al. [60] suggested a positive correlation between WMH burden and the risk of recurrence of spontaneously hypertensive ICH, and the more serious the lesion, the more significant the correlation between them. Moreover, WMH of different lesion sites differentially affected cognition. Some studies have found that WMH of the frontal lobe mainly affects executive function; parietal-temporal WMH near the posterior horn mainly affects memory; and WMH of the deep white matter (including the corticospinal tract) significantly influences response speed [61,62]. Hypertension, type 2 diabetes, and smoking are considered the main risk factors for WMH [63].
Convex subarachnoid hemorrhage and cSS
CAA-related ICH is often accompanied by cSAH, another important CAA imaging marker. In general, cSAH refers to non-traumatic, non-aneurysmal SAH that only involves the sulcus of the cerebral cortex and does not involve the adjacent brain parenchyma or extend to the interhemispheric fissure, basal cistern, or ventricle [64]. It may be a characteristic manifestation of decreased vascular elasticity [65]. Conversely, cSS refers to the deposition of hemosiderin in the pia mater and/or spinal cord, which is generally considered to be the conversion of cSAH to subcortical hemosiderin deposition after weeks to months.
The FLAIR sequence has a high sensitivity and specificity for the diagnosis of acute cSAH. FLAIR mainly showed high signal intensity in one or more cerebral sulci in the convex cortex and low signal intensity in the GRE sequence, whereas cSS showed low signal intensity on both sides of the supratentorial sulcus on the T2*GRE sequence, showing a track appearance, but no corresponding high signal on FLAIR [66]. cSAH/cSS can serve as important imaging biomarkers of early recurrent CAA-ICH. Further in-depth research is needed on the relevant pathophysiological mechanisms and imaging manifestations to provide more evidence for the clinical diagnosis of CAA-ICH.
Enlarged perivascular space
PVS is the physiological space around small blood vessels, passing through the brain parenchyma from the subarachnoid space, and is considered to be part of the lymphatic system. It is an important channel for removing metabolic waste from the brain and maintaining the dynamic balance of fluid circulation [67-69]. Affected by age, sex, cardiovascular risk factors, APOE, head trauma, sleep disorders, and other magnetic resonance imaging (MRI) markers such as WMH [70-72], dysfunctional PVSs lead to the accumulation of toxins and physiological metabolites produced in the brain, resulting in further pathological damage and EPVS. It is generally believed that the formation of EPVS is related to the limitation of Aβ clearance.
EPVS can be quantified using high-resolution MRI. Common sites of EPVS include the basal ganglia, subcortical white matter, midbrain, and hippocampus, among which the central semiovale, basal ganglia, and midbrain are the three characteristic sites [73]. In view of its differential distribution, the EPVS signal on MRI was similar to that of the cerebrospinal fluid, and there was no obvious space-occupying or enhancement effect. Different sections are differently shaped, mainly showing clear boundaries and round, oval, or linear structures, with a maximum diameter of ≤3 mm [74]. These showed low signals on T1-weighted imaging (T1WI) sequences, high signals on T2WI, and low signals on FLAIR sequences, and were often accompanied by a perforating artery [75].
Cerebral microinfarcts
CMIs cannot be seen with the naked eye. Clear, defined lesions of cell death or tissue necrosis in the brain can be observed under a microscope, and sometimes cavities can be observed. The pathological manifestations are similar to those of common ischemic infarctions [76]. Corrada et al. [77] first described the pathological characteristics of CMI, namely, pale lesions, loss of vulnerable cells, proliferation of glial cells, and the presence of macrophages. CMI is considered to be closely related to intracranial atherosclerosis, arteriosclerosis or occlusion, stroke history, CAA, cerebral hypoperfusion, heart disease, etc.
Owing to the small size of microinfarctions, tracking them using conventional MRI is difficult. In recent years, with the continual development and innovation of imaging techniques, CMI usually shows cortical high-signal intensity on 7-T MRI with an ultra-high field strength in vitro on FLAIR and T2 sequences, whereas it shows low-signal intensity on T1 sequences, which is different from the pattern exhibited by PVS lesions of ≤3 mm size [78]. In addition, 3-T MRI can also detect the presence of CMI, but its sensitivity is very low compared to that of 7-T MRI (30%-40%) [79]. The results of the Medea-7T study showed that the frequency of CMI on 7-T MRI was similar in patients with vascular diseases and from memory clinics [80]. Microinfarction can be observed throughout the brain, and their distribution and number are closely related to the type and progression of dementia. Therefore, CMI may be an independent risk factor for CAA-associated cognitive impairment [81].
Diagnostic criteria for CAA and CAA-ri
Diagnosis of CAA
Currently, the diagnostic criteria for CAA are based on cohort study data. To date, the diagnostic criteria for CAA mainly include the Boston, Edinburgh, and CAA-ri diagnostic criteria. Among these, the most recognized Boston diagnostic criteria were updated to version 2.0. The evolution of the three versions of the diagnostic criteria is shown in Table 1 [82-84]. Patients who are unable to complete MRI examinations based on ICH indicated by computed tomography can be evaluated according to the presence or absence of SAH, finger hemorrhage, and APOE. Therefore, the Edinburgh diagnostic criteria were established (Table 2) [85].
Diagnostic criteria for CAA-ri
In 2011, based on the clinical manifestations and MRI results of patients, clinical diagnostic criteria for CAA-ri were proposed for the first time and were divided into confirmed CAA-ri and probable CAA-ri [86]. In 2016, Auriel et al. [87] updated the clinical imaging diagnostic criteria for CAA-ri based on the 2011 criteria. The evolution of the two sets of diagnostic criteria is shown in Table 3. In addition, pathological results confirmed that the sensitivity and specificity of the diagnostic criteria for probable CAA-ri were 82% and 97%, respectively, whereas those for possible CAA-ri were 82% and 68%, respectively [87].
Treatment for CAA and CAA-ri
To date, no specific treatment method is available for CAA and CAA-ri. Long-term management of patients with CAA mainly focuses on reducing the incidence and recurrence of cerebral hemorrhage and preventing dementia. The treatment method for CAA-related acute cerebral hemorrhage is the same as that used for other spontaneous cerebral hemorrhages. For patients with CAA and lobar cerebral hemorrhage, APT should be avoided as much as possible to reduce the risk of recurrent cerebral hemorrhage.
The American Heart Association/American Stroke Association recommends that ACT be avoided as much as possible for patients with previous CAA-ICH; however, patients with no history of cerebral hemorrhage can continue oral ACT [88]. In addition, blood pressure control in the acute stage of CAA-related ICH is closely associated with patient prognosis. Data from a systematic review and meta-analysis published by Moullaali et al. [89] showed that compared with placebo/guideline treatment, active/intensive antihypertensive intervention within 7 days after acute ICH significantly reduced the size of absolute/relative intracerebral hematoma, although it did not improve the patients’ functional prognosis. However, Rehni et al. [90] found that treatment with red blood cell-derived microparticles in a rat model of ICH significantly limited the expansion of hematoma and improved the prognosis of ICH. A study using a mouse model of ICH also found that meningeal lymphangiogenesis and lymphatic drainage increased on days 10-14 after ICH and lasted for at least 2 months after ICH. Early enhancement of the meningeal lymphatic function can promote hematoma absorption and reduce hematoma volume, thereby improving mouse behavior. These studies provide a new research direction for CAA-ICH diagnosis and treatment, which will improve patient prognosis by controlling blood pressure and promoting hematoma absorption [91].
In recent years, research has shifted from CAA-related treatments to immunotherapies. Xiong et al. [92] showed that the specific binding of anti-APOE antibody HAE-4 to low-fat APOE in the core of the plaque reduced amyloid protein deposition, while simultaneously inhibiting reactive microglia and astrocytes in the cortex, thus improving CAA-induced cerebral vascular dysfunction. An experiment on Tg-SwDI mice confirmed that fruquintinib, a highly selective antibody against vascular endothelial growth factor receptor (VEGFR), reduced VEGFR accumulation and controlled inflammation and neuronal death, thereby reducing Aβ deposition in the cerebral cortex and hippocampus of mice, effectively treating CAA [93].
Moreover, given that previous research has suggested that CAA-ri is a special autoimmunity-mediated subtype of CAA, immunotherapy is considered a key treatment for CAA-ri. Regenhardt et al. [94] reported favorable results from immunosuppressive therapy including corticosteroids and cyclophosphamide for CAA-ri, with 94% patients showing improved clinical symptoms and 86% with improved imaging findings; thus, immunosuppressive therapy reduced the risk of CAA-ri recurrence.
Conclusion and future prospects
In the past 10 years, research on CAA has been ongoing and productive. Understanding the pathophysiology of CAA is expected to help provide systematic and personalized diagnosis and treatment programs for the control of blood pressure, medical and surgical intervention for hematoma, and discover emerging immunotherapies. Additionally, continual innovation in new imaging methods and testing of cerebrospinal fluid and genes will forge the path to a more accurate diagnosis and treatment route for CAA, especially CAA-ri. Thus will lower patient mortality and improve their quality of life.