Cerebral Amyloid Angiopathy: Emerging Concepts
Article information
Abstract
Cerebral amyloid angiopathy (CAA) involves cerebrovascular amyloid deposition and is classified into several types according to the amyloid protein involved. Of these, sporadic amyloid β-protein (Aβ)-type CAA is most commonly found in older individuals and in patients with Alzheimer's disease (AD). Cerebrovascular Aβ deposits accompany functional and pathological changes in cerebral blood vessels (CAA-associated vasculopathies). CAA-associated vasculopathies lead to development of hemorrhagic lesions [lobar intracerebral macrohemorrhage, cortical microhemorrhage, and cortical superficial siderosis (cSS)/focal convexity subarachnoid hemorrhage (SAH)], ischemic lesions (cortical infarction and ischemic changes of the white matter), and encephalopathies that include subacute leukoencephalopathy caused by CAA-associated inflammation/angiitis. Thus, CAA is related to dementia, stroke, and encephalopathies. Recent advances in diagnostic procedures, particularly neuroimaging, have enabled us to establish a clinical diagnosis of CAA without brain biopsies. Sensitive magnetic resonance imaging (MRI) methods, such as gradient-echo T2* imaging and susceptibility-weighted imaging, are useful for detecting cortical microhemorrhages and cSS. Amyloid imaging with amyloid-binding positron emission tomography (PET) ligands, such as Pittsburgh Compound B, can detect CAA, although they cannot discriminate vascular from parenchymal amyloid deposits. In addition, cerebrospinal fluid markers may be useful, including levels of Aβ40 for CAA and anti-Aβ antibody for CAA-related inflammation. Moreover, cSS is closely associated with transient focal neurological episodes (TFNE). CAA-related inflammation/angiitis shares pathophysiology with amyloid-related imaging abnormalities (ARIA) induced by Aβ immunotherapies in AD patients. This article reviews CAA and CAA-related disorders with respect to their epidemiology, pathology, pathophysiology, clinical features, biomarkers, diagnosis, treatment, risk factors, and future perspectives.
Introduction
Cerebral amyloid angiopathy (CAA) involves cerebrovascular amyloid deposition. CAA was described as a cause of normotensive cerebral hemorrhage in older individuals or also described in studies focusing on its close association with Alzheimer's disease (AD). The earlier terms to describe CAA include "drüsige Entartung der Arterien und Kapillaren,"1 "aniopathie dyshorique,"2 and "angiopathie congophile."3 The terms "drüsige Entartung" and "angiopathie dyshorique" represent a peculiar form of CAA showing perivascular plaque-like structures. Currently, CAA has been used as a general term to describe cerebrovascular amyloid deposition or cerebrovascular amyloidosis.4
CAA is classified into several types according to the amyloid protein involved (Table 1). So far, seven amyloid proteins have been reported in CAA including amyloid β-protein (Aβ), cystatin C (ACys), prion protein (APrP), ABri/ADan, transthyretin (ATTR), gelsolin (AGel), and immunoglobulin light chain amyloid (AL); among these, sporadic CAA of the Aβ type is most commonly found in older individuals as well as in patients with AD [see review5].
Classification of cerebral amyloid angiopathy (CAA)
This article reviews sporadic Aβ-type CAA and CAA-related disorders with respect to their epidemiology, pathology, pathophysiology, clinical features, biomarkers, diagnosis, treatment, risk factors, and future perspectives.
Epidemiology, pathology, and pathophysiology of CAA
The prevalence of CAA increases with age,6,7 and CAA occurs in approximately half of elderly individuals.6 CAA is commonly observed in AD with a prevalence of about 80%-90%.6,8 CAA-related lobar intracerebral hemorrhage (ICH), identified via a nationwide survey in Japan, increases with age and presents with female predominance.9
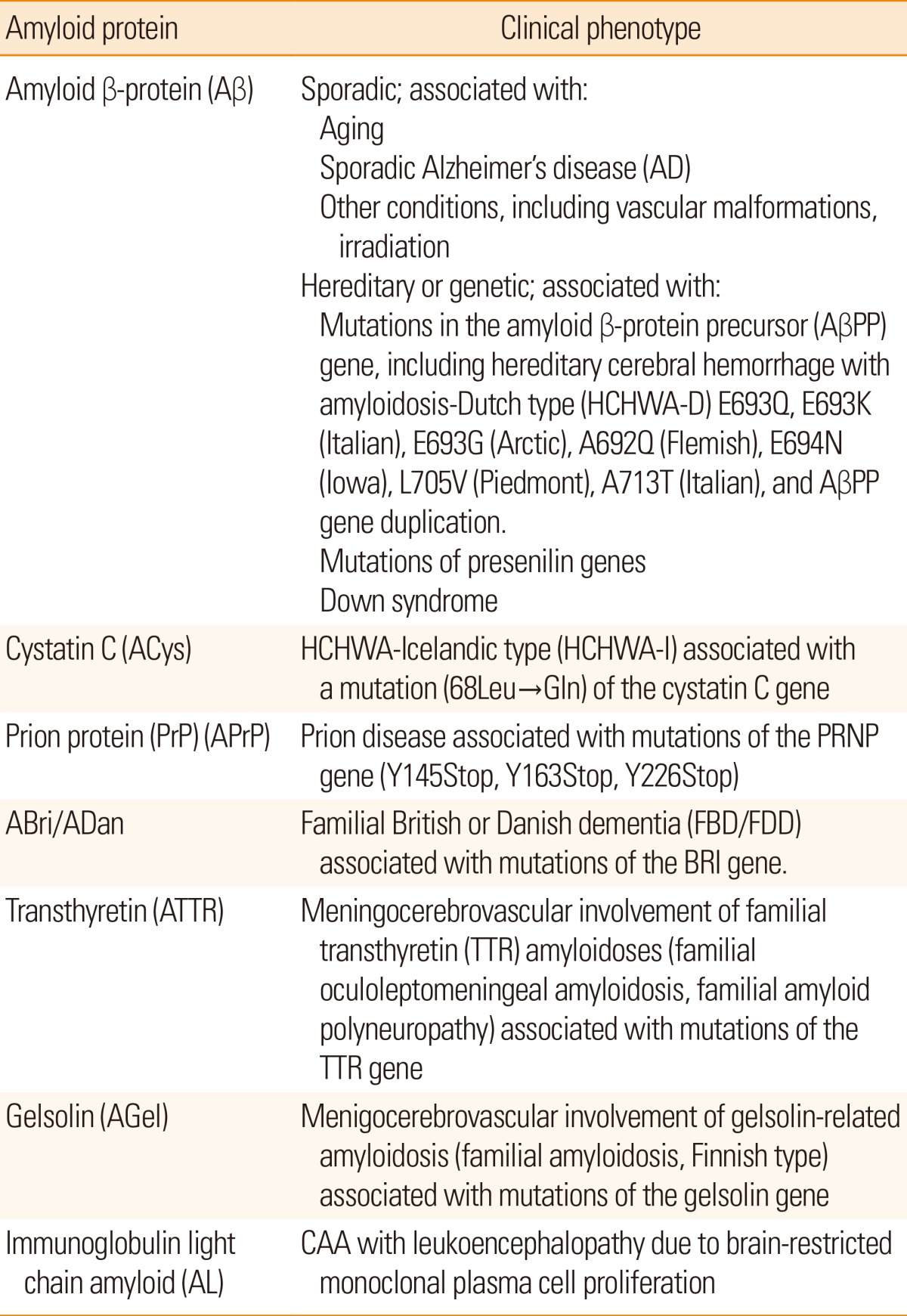
Pathologically, CAA is observed mainly in the leptomeningeal and cortical vessels of the cerebral lobes and cerebellum. For the distribution of CAA, the occipital lobe is preferentially affected,6 whereas CAA is uncommon in the basal ganglia, thalamus, brainstem, and white matter. In mild CAA, a small proportion of the leptomeningeal and superficial cortical vessels are affected with amyloid deposition. In severe CAA, most small arteries and arterioles are affected with marked amyloid deposition. Medium-sized leptomeningeal arteries show amyloid deposition in the outer regions of the media to the adventitia. Vessel walls of the small arteries and arterioles are often totally replaced by amyloid deposits, except for the endothelial cells. Electron microscopically, amyloid fibrils are focally deposited within the outer basement membrane of the vessels during the initial stage of CAA.10 Later, a large amount of amyloid fibrils accumulate with degeneration of smooth muscle cells of the media (Figure 1A). Amyloid deposits in capillaries and, occasionally, in arterioles or small arteries appear to infiltrate the surrounding parenchymal tissue, and accompany dystrophic neurites forming plaque-like structures (drüsige Entartung1/angiopathie dyshorique2). Recently, CAA in capillaries has been referred to as "capillary CAA (CAA-Type 1)," to distinguish it from non-capillary CAA (CAA-Type 2).11 Severe CAA is associated with vasculopathies including duplication ("double-barrel" lumen), obliterative intimal changes, hyaline degeneration, microaneurysmal dilatation, and fibrinoid necrosis (Figure 1B, C).12,13,14 These CAA-associated vasculopathies are the pathological basis of CAA-related cerebrovascular disorders. In particular, fibrinoid necrosis is closely associated with CAA-related hemorrhage.12,13,14
CAA and CAA-associated vasculopathies. Massive amyloid deposition of amyloid fibrils with degeneration of smooth muscle cells in the media (A). Microaneurysmal dilatation (arrow) with fibrinoid necrosis (*) (B). Thickening of the intima (arrow) and double barreling of vascular walls (arrowheads) (C). (A, electron micrograph, bar=1 µm; B, Congo red, original magnification 110×; C, Congo red, original magnification 170×).
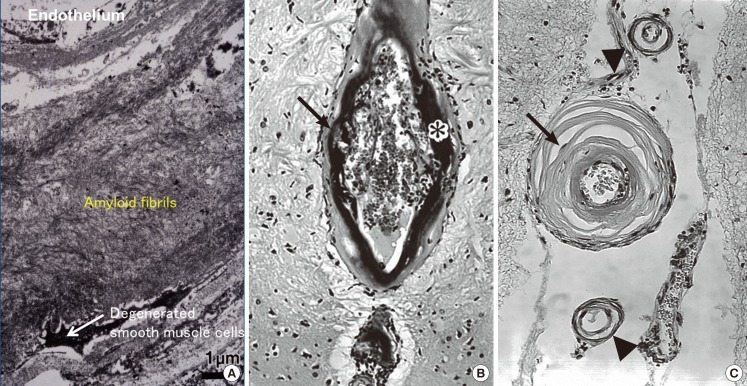
Aβ is cleaved from the β-amyloid precursor protein (AβPP) by β-secretase and γ-secretase. Heterogeneity of the C-terminal is present, and the length of Aβ in senile plaques is mainly 42-43 residues (Aβ42), while that of cerebrovascular Aβ (CAA) is mainly 39-40 residues (Aβ40) (Figure 2).15,16 Recent studies suggest that Aβ in CAA is derived from the brain [see review5]; after release from neurons, Aβ42 easily aggregates and deposits in the brain parenchyma as senile plaques; whereas, Aβ40 does not aggregate as easily as Aβ42, and is transported, through periarterial interstitial fluid drainage pathways, to blood vessels for clearance. In this process, Aβ40 aggregates on vascular basement membranes, as first proposed by Weller.17
Immunohistochemistry of adjacent brain sections with antibodies to Aβ40 (A) and Aβ42 (B). Positive immunoreactivity to Aβ40 is mainly observed in vessel walls (CAA) (A), whereas Aβ42 immunoreactivity is mainly observed in the brain parenchyma (senile plaques) (B).
The pathophysiology of Aβ-type CAA is shown in Figure 3.18 CAA-associated vasculopathies lead to development of hemorrhagic lesions [lobar intracerebral macrohemorrhage, cortical microhemorrhage, and cortical superficial siderosis (cSS)/focal convexity subarachnoid hemorrhage (SAH)], ischemic lesions (cortical infarction and ischemic changes of the white matter), and encephalopathies that include subacute leukoencephalopathy caused by CAA-related inflammation/angiitis (Figure 1). In addition, CAA is related to dementia through AD pathology and CAA-related vascular lesions (Figure 1). Thus, CAA is related to dementia, stroke, and encephalopathies.
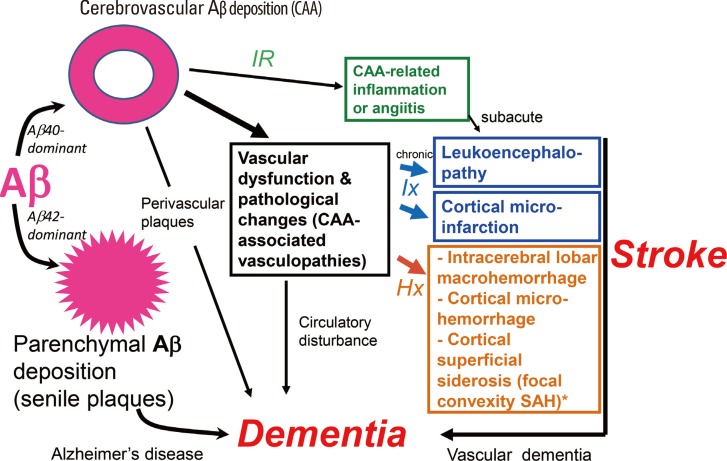
Pathophysiology of cerebral amyloid angiopathy (CAA)-related disorders. Aβ shows parenchymal (senile plaques) or vascular deposition (CAA) depending on dominance of Aβ42 or Aβ40, respectively. CAA is related to stroke and dementia. Hx, hemorrhagic events; Ix, ischemic events; IR, immune reaction against Aβ.
Clinical aspects of CAA-related disorders
Clinical features
Intracerebral hemorrhages
Symptomatic lobar intracerebral hemorrhages (ICH)
CAA is significantly associated with lobar intracerebral hemorrhage (ICH) (Figure 4A), but not with deep ICH,19 because sporadic Aβ-type CAA is commonly found in the meningeal and cortical vessels of cerebral and cerebellar cortices, and rarely in those of the deep gray matter including basal ganglia, thalamus, and brainstem.6 Using the SMASH-U system (structural lesion, medication, amyloid angiopathy, systemic/other disease, hypertension, undetermined) as a pathogenetic classification system for ICH, CAA-related ICH was noted in 20% of ICH cases in the Helsinki ICH study,20 and 12% of ICH cases in the National Taiwan University Hospital Stroke Registry.21 CAA-related ICH was the second most common cause of ICH following hypertensive angiopathy in these studies.20,21 The incidence of lobar ICH in the elderly has been increasing recently, in which CAA is strongly implicated.22 CAA-related lobar ICH is often multiple and recurrent, and clinical manifestations include motor paresis, disturbance of consciousness, abnormalities in higher brain functions, such as aphasia, visual loss, with headache at the acute stage, and dementia and seizures during chronic stages.9 Headache with meningeal signs is likely caused by subarachnoid hemorrhage (SAH) accompanying lobar ICH.23,24,25
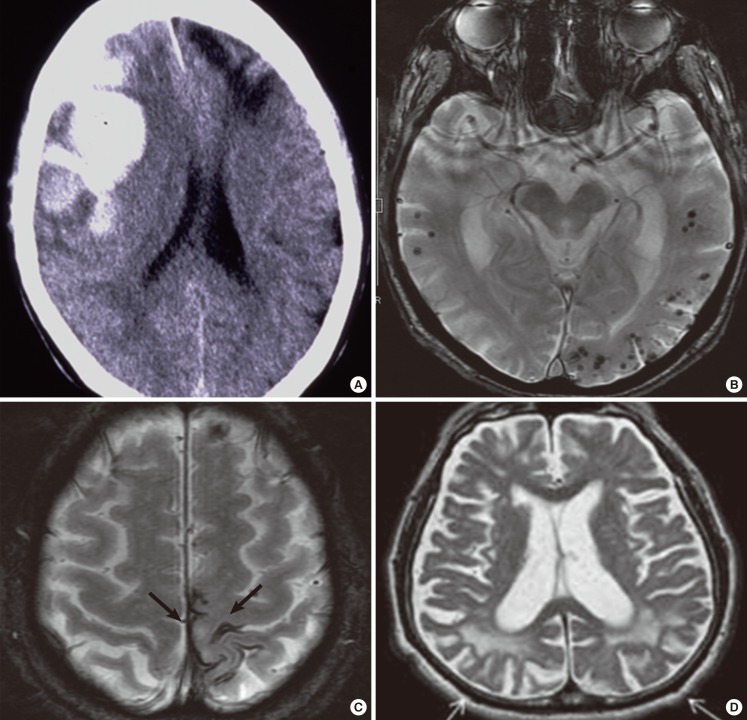
Imaging findings of CAA-related hemorrhages and white matter lesions. Fresh (arrow) and old (arrowhead) lobar macrohemorrhages in the frontal lobes on CT (A). Cortical microhemorrhages with lobar distribution (B) and focal subarachnoid hemorrhages (superficial siderosis) (C) on gradient echo T2*-weighted MRI. Posterior distribution of white matter hyperintensities (arrows on T2-weighted MRI) (D).
Cortical microhemorrhages
Cerebral microhemorrhages (microbleeds) were noted in 16.7%-32% of AD patients, which is higher than in the general population (5%-6%), when examined by gradient-echo T2* MRI,26,27,28,29,30 and in 78% of patients with AD or mild cognitive impairment (MCI) on ultra-high field strength 7T MRI.31 Microhemorrhages were found in 47.4% of patients with pathologically confirmed CAA cases.32 CAA-related microhemorrhages are frequently lobar in distribution (Figure 4B). Studies with amyloid positron emission tomography (PET) using 11C-Pittsburgh Compound B (PiB) reported that lobar microhemorrhages are frequently found in PiB-positive healthy subjects as well as patients with AD or mild cognitive impairment (MCI),33 and that microhemorrhages preferentially occur in local regions of concentrated amyloid deposits.34 New hemorrhages occur preferentially at the sites of increased amyloid deposits, suggesting that amyloid imaging may be useful to predict future CAA-related hemorrhages.35 Patients with lobar microhemorrhages are at considerable risk of future symptomatic lobar ICH.36
Cortical superficial siderosis/focal convexity subarachnoid hemorrhages
CAA is a frequent cause of cSS/focal convexity SAH (Figure 4C), a subtype of nonaneurysmal SAH, in patients over the age of 60 and in those with AD.32,37,38 Notably, cSS is closely associated with microhemorrhages in lobar locations in the general population.39 Moreover, cSS occurs with high prevalence in CAA and is found in 60.5% of pathologically confirmed cases.32,40 Interestingly, cSS tends to occur in individuals with relatively fewer cortical microhemorrhages, suggesting differences in vasculopathic changes between CAA-related microhemorrhages and cSS.41 Furthermore, cSS is associated with an increased future risk of symptomatic lobar ICH.40,42 The prevalence of cSS is higher in patients with AD or cognitive impairment than in the general population.43,44 Interestingly, cSS has been reported to present with transient focal neurological episodes (TFNEs); TFNEs are a clinical marker of cSS and may be caused by cSS through cortical spreading depression or focal seizure activity.45,46,47,48,49 Anti-platelet or anticoagulant therapies based on the misdiagnosis of CAA-related TFNE as a transient ischemic attack may induce CAA-related ICH.
White matter disease and cortical infarction
CAA-related cerebral hypoperfusion or occlusive small-vessel disease may cause progressive white matter lesions and cortical microinfarcts. Patients with CAA-related ICH exhibit occipital dominant white matter hyperintensities (WMHs) on MRI (Figure 4D), compatible with predilection of CAA pathology for posterior brain regions.50 A posterior distribution of WMH on MRI is associated with the presence of CAA pathology.51 Amyloid burden in non-demented CAA subjects correlated with WMH volumes.52 Acute or subacute subclinical ischemic infarcts are common in CAA-related ICH.53,54,55 As a result of the difficulty in detection (see below), CAA as a cause of cortical microinfarcts is underrecognized.
Dementia
CAA with hemorrhagic/ischemic vascular lesions or AD pathology may be associated with dementia during its clinical course,56 although dementia is found only in a subgroup of patients with CAA-related ICH at the onset of the initial ICH.9,57 Dementia was noted in 74% of individuals with severe CAA at autopsy.56 Moderate-to-very severe CAA is associated with impaired performance in specific cognitive domains, most notably perceptual speed, which is separate from the effect of AD pathology.58
CAA-related inflammation
CAA is commonly associated with immune reactions of monocyte/macrophage-lineage cells.59 CAA can be accompanied by marked inflammation (CAA-related inflammation/angiitis), presenting with subacute leukoencephalopathy that is responsive to immunosuppressive therapies.59,60,61,62,63 The clinical symptoms include subacute cognitive impairment or seizure, which are different from those of hemorrhagic stroke in noninflammatory CAA. Inflammatory CAA is also characterized by asymmetric T2-hyperintense white matter lesions on MRI.59,60,61,62,63 CAA-related inflammation is associated with an increase in anti-Aβ antibodies in cerebrospinal fluid (CSF).64,65 CAA-related inflammation/angiitis shares pathophysiology with amyloid-related imaging abnormalities (ARIA) induced by Aβ immunotherapies, as discussed below.
Biomarkers and diagnosis
Imaging markers
CT/MRI
Cranial CT is an option for macrohemorrhages; however, sensitive MR imaging techniques, such as gradient-echo T2* imaging and susceptibility-weighted imaging, are useful for detecting microhemorrhages and cSS.66 A posterior distribution of WMH is a possible marker of CAA.51 Although acute or subacute cortical or subcortical infarctions can be recognized in CAA on diffusion-weighted images,53 cortical microinfarcts are often undetected on MRI because of their limited size.31 Ultra-high strength clinical MR scanners with a higher spatial resolution may enable their detection.67 CT angiography spot signs, suggestive of an acute bleeding process, are associated with warfarin use and ICH volume in both CAA-related lobar ICH and arteriolosclerotic deep ICH.68 Enlarged perivascular spaces in the centrum semiovale are associated with CAA, suggesting an imaging marker for CAA.69,70,71
Amyloid PET
Amyloid imaging with the PET ligand, PiB, revealed an increase of PiB binding that often shows greater occipital uptake in CAA-related ICH (Figure 5).75,76,77 Microhemorrhages are associated with PiB retention,33,34 and new CAA-related hemorrhages preferentially occur at sites of PiB retention.35 As a frequent occurrence of high PiB uptake in healthy elderly individuals reflects incipient AD, PiB PET has low specificity for CAA; however, a negative PiB scan rules out CAA with excellent sensitivity.78 Amyloid imaging with such PET ligands as PiB cannot discriminate vascular from parenchymal deposition or Aβ from other amyloid proteins; reagents specific for vascular Aβ deposition are favorable candidates for amyloid imaging specific for Aβ-type CAA.79

Amyloid positron emission tomography (PET) using 11C-Pittsburgh Compound B (PiB) (A) with gradient echo T2*-weighted MRI (B) in a non-demented patient with multiple CAA-related intracerebral hemorrhages and disseminated cortical superficial siderosis. The left parietal region with an old intracerebral hemorrhage shows a relative scarcity of PiB uptake.
Biochemical markers
A significant decrease in cerebrospinal fluid (CSF) levels of Aβ40 as well as Aβ42 was reported in patients with probable CAA, which suggests trapping of Aβ40 and Aβ42 in the cerebral vasculature. Furthermore, CSF levels of total tau and phosphorylated tau are higher in patients with probable CAA than in controls, but lower than in AD.80,81 It was reported that patients with CAA-related ICH exhibit higher plasma levels of Aβ42, Aβ40, and specific Aβ fragments.82 The presence of anti-Aβ autoantibodies in CSF is a marker of CAA-related inflammation.64,65
Diagnosis
The Boston criteria were proposed for the diagnosis of CAA-related ICH (Table 2), and high diagnostic accuracy was reported with a small pathologic series.83 As hypertension may cause lobar ICH,23 exclusion of advanced hypertension may increase the specificity of the diagnosis of CAA-related ICH; however, the diagnostic sensitivity is decreased,9 because CAA may coexist with hypertension.

When cSS was incorporated into the classic Boston criteria (the modified Boston criteria) (Table 2), the sensitivity increased from 89.5% to 94.7%, and the specificity was 81.2% for both the classic and modified criteria.32
Current treatment and prognosis
Currently, no disease-modifying therapies are available for CAA. As for neurosurgery for CAA-related ICH, uncontrollable peri- and post-operative hemorrhages have been reported previously; however, recent studies suggest that neurosurgical procedures, especially hematoma evacuation, can be performed more safely than previously expected.84 In our study involving a nationwide survey in Japan,9 neurosurgical procedures were performed without uncontrollable intra-operative or post-operative hemorrhage in 97.1% of patients. CAA-related lobar ICH recurred in 31.7% of patients during the average 35.3-month follow-up period. The mean interval between ICHs was 11.3 months and the case fatality rate was 12.2% at 1 month and 19.5% at 12 months after initial ICH. Subacute leukoencephalopathy associated with CAA-related inflammation or angiitis was reported to respond to immunosuppressive treatment.61,62,63
Risk factors of CAA and CAA-related disorders
Aging and AD are established risk factors of CAA;5 besides these, genetic and non-genetic risk factors have been reported for CAA and CAA-related disorders.
Genetic risk factors
Apolipoprotein E gene
In addition to hereditary cases of AD/CAA associated with mutations of the AβPP or presenilin genes [see review5], the ApoE gene (ApoE) has been reported to be risk factor for sporadic CAA as well as AD; the ε4 allele for CAA itself, and the ε2 allele for CAA-related ICH85,86,87 and hematoma expansion.88 Carriers of the ApoE ε2 or ε4 allele, particularly, of the ApoE ε2/ε4 genotype were associated with early recurrence of lobar ICH in patients who survived a lobar ICH.89 The ApoE ε4 allele constitutes a risk factor for capillary CAA (CAA-Type 1),11 CAA-related inflammation,63 and brain microhemorrhages.90 Furthermore, ApoE ε2 was overrepresented in patients with cSS.41 The presence of CAA in head injury cases was significantly associated with the ApoE ε4 allele, suggesting an interaction between gene and environment in the development of CAA.91
Other genetic factors
Transforming growth factor (TGF)-β1 is another CAA-related gene reported by more than one research group.92,93 In addition, CAA was reported to be associated with other gene polymorphisms, including the presenilin 1 (PS1), α1-antichymotrypsin (ACT), neprilysin, low-density lipoprotein-receptor related protein (LRP-1), and angiotensin-converting enzyme (ACE) genes.8,94,95,96,97,98,99,100 Furthermore, a CR1 gene polymorphism, reported to increase the risk of AD, was also reported to be associated with increased risk of CAA-ICH and CAA.101
Non-genetic risk factors
Hypertension
Lowering of blood pressure reduced the risk of CAA-related ICH, suggesting that high blood pressure could be a factor inducing ICH in patients with CAA.102 Hypertension could contribute to progression of CAA-associated vasculopathies, particularly fibrinoid necrosis, leading to the development of CAA-related ICH. It was reported that the prevalence of hypertension was significantly higher in autopsy-confirmed CAA patients with ICH than in those without ICH, suggesting a role for hypertension in the development of CAA-related ICH.13
Thrombolytic, anticoagulation, and antiplatelet therapies
There is increasing evidence that CAA could be a risk factor for ICH with thrombolytic therapies for acute myocardial infarction, pulmonary embolism, or ischemic stroke, and for ICH with warfarin therapies.103,104 Thrombolytic or anticoagulation therapies in patients with micorhemorrhages are a potential risk factor for ICH.36,105,106 The ApoE ε2 and ε4 alleles were reported as strong risk factors for lobar warfarin-related ICH; this association was considered to be mediated by the effect of ApoE ε variants on CAA.107 The use of anti-platelet drugs, such as aspirin, is associated with the presence of microhemorrhages, and with strictly lobar microhemorrhages suggestive of CAA.108 In a systematic review of clinical trials with aspirin in AD, ICH was noted in 3.2% of patients in the aspirin group, but in 0% of the control group, suggesting that aspirin use in AD might increase the risk of ICH. A possible explanation for the increased risk of ICH could be CAA in AD.109
Anti-amyloid therapies
Clinical and experimental studies of Aβ immunotherapies for AD have reported CAA-related complications. In clinical trials with Aβ42 immunization for AD (AN1792, Elan), immunized patients showed a significantly greater frequency of CAA, cortical microhemorrhages, and microvascular lesions than unimmunized AD controls, although the longest living had a virtually complete absence of both plaques and CAA.110 A CAA-related macrohemorrhage was reported in an AD patient in phase 2a of AN1792.111 The findings suggest that Aβ immunization resulted in solubilization of Aβ42 in plaques, which migrates out of the brain parenchyma via perivascular interstitial drainage pathways, causing an increase in CAA and CAA-related hemorrhages.110 Menigoencephalitis occurred in 6% of patients treated in the AN1792 trial,111 and perivascular infiltration of lymphocytes was observed around vessels with CAA.112 Furthermore, in a phase 2 trial of bapineuzumab, a humanized monoclonal anti-Aβ antibody, amyloid-related imaging abnormalities (ARIA) were reported: vasogenic edema and sulcal effusions (ARIA-E) in 17%, and microhemorrhages and hemosiderin deposits (ARIA-H) in 47% of the patients with ARIA-E,113 suggesting that Aβ immunotherapy resulted in changes in permeability and disruption of CAA-affected vessels. Vasogenic edema was noted in 2 of 2,762 patients with AD at the baseline of the clinical trial; findings were compatible with CAA-related inflammation.114 As discussed above, CAA-related inflammation is associated with the presence of anti-Aβ autoantibodies in CSF.64,65 Further studies are required to elucidate the link between Aβ immunotherapy-induced ARIA and CAA-related inflammation.
Future perspectives of prediction, prevention, and therapies for CAA and CAA-related disorders
Biomarker findings and risk factors for CAA and CAA-related disorders are summarized in Table 3. A number of studies indicate that the presence of one or more biomarkers plus one or more risk factors suggest the future development of CAA-related disorders. Further prospective studies will establish methods for prediction of CAA-related disorders. To develop preventive and therapeutic methods for CAA and CAA-related disorders, their molecular pathogenesis needs to be further elucidated. The molecular pathogenesis of Aβ deposition in cerebral blood vessels is discussed in detail elsewhere [see review5].
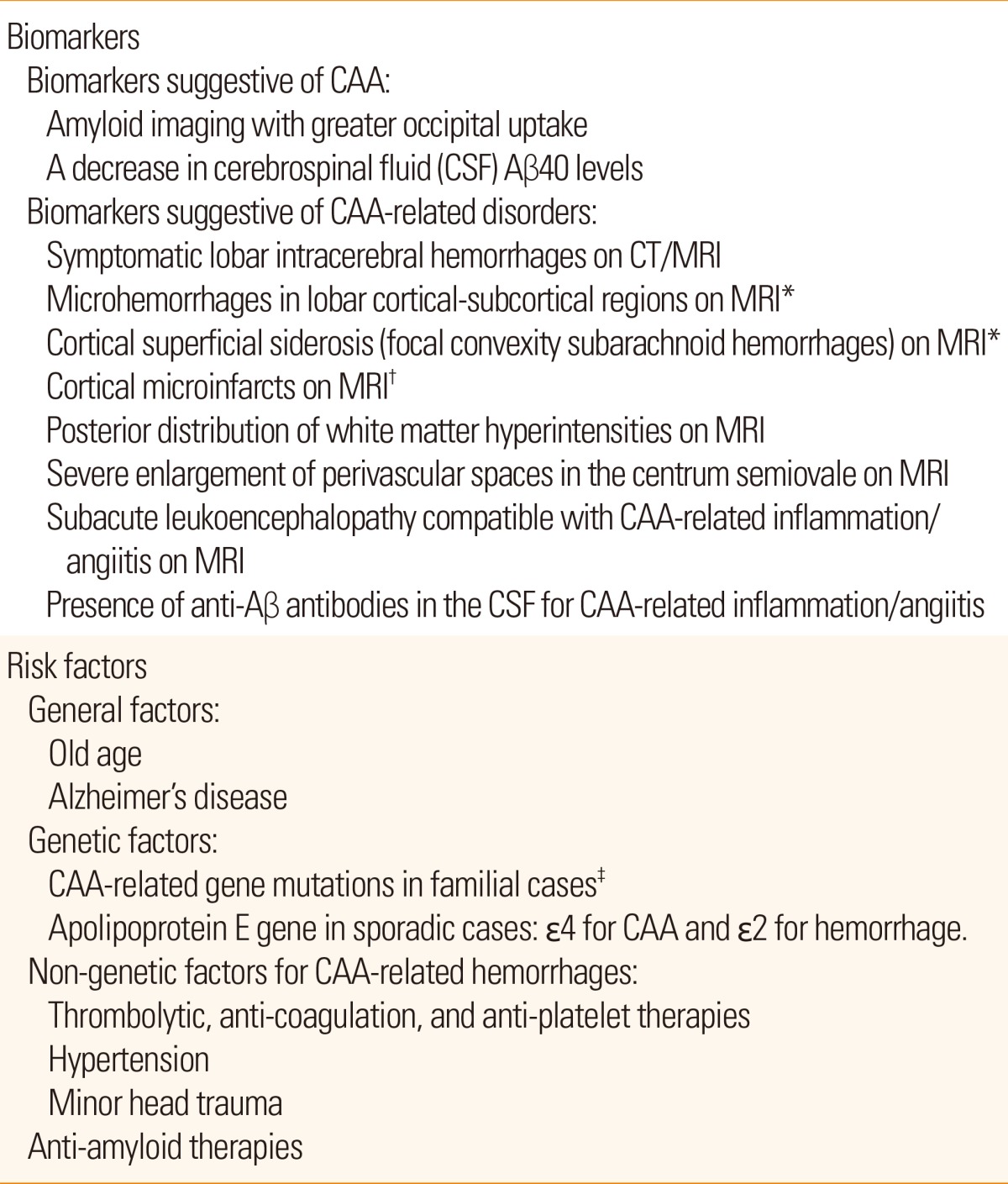
Biomarkers and risk factors for cerebral amyloid angiopathy (CAA) and CAA-related disorders
CAA is not necessarily associated with CAA-related disorders. The pathogenesis of CAA-related disorders consists of two steps: (1) cerebrovascular amyloid deposition and (2) vascular injury including disruption, occlusion, and permeability changes. The cerebrovascular amyloid deposition (Step 1) is the primary target of prevention and therapy. Anti-amyloid therapies for CAA are under development including ponezumab, a humanized monoclonal antibody that binds specifically to the carboxyl terminus of Aβ40.115 Ponezumab was originally developed for AD immunotherapy, and an acceptable safety profile has been suggested for ponezumab in clinical trials for AD.116,117,118 Currently, ponezumab has been applied to CAA. A phase 2, randomized, double-blind, placebo-controlled trial is ongoing to evaluate the safety, tolerability, pharmacokinetics, and efficacy of ponezumab (PF-04360365) in adult patients with probable CAA-related hemorrhages (NCT01821118).119
Vascular injury secondary to cerebrovascular amyloid deposition (Step 2) is an important target for prevention of CAA-related disorders. Risk factors and pathogenic mechanisms underlying vascular injury leading to CAA-related hemorrhages are discussed below. For CAA-related hemorrhages, several non-genetic risk factors have been reported and it is recommended to avoid them. These include thrombolytic, anticoagulation, and anti-platelet therapies, hypertension, and minor head trauma in patients with positive biomarkers of CAA and CAA-related disorders (Table 3). Pathogenic mechanisms underlying the damage and rupture of CAA-affected vessels remain to be determined. Pathologically, severe CAA is associated with vasculopathies including loss of smooth muscle cells, duplication ("double-barrel" lumen), obliterative intimal changes, hyaline degeneration, microaneurysmal dilatation, and fibrinoid necrosis of the vessel walls (Figure 1).12,13,14 Of these, fibrinoid necrosis, in particular, is closely linked to CAA-related hemorrhage.12,13,14 In addition, even in cases without obvious inflammation or angiitis, CAA-affected vessels are associated with an increase in and activation of monocyte/macrophage lineage cells representing immune reactions against vascular amyloid deposition,59 which would damage the vessel walls. Aβ deposits in blood vessel walls may trigger release of inflammatory components, activation of the complement system, oxidative stress, alterations in blood brain barrier permeability, formation of ion-like channels, and cell toxicity.120 The involvement of matrix metalloproteinases in the degradation of extracellular matrix proteins was suggested in an experimental model and in human brains exhibiting CAA-related ICH.121,122 Soluble Aβ oligomers, which are more neurotoxic than insoluble Aβ fibrils, may damage vessel walls affected with CAA. There is increased evidence that ApoE ε2 is associated with CAA-related ICH and vessel fragility;87,88,123,124 however, ApoE-related pathogenic mechanisms remain to be elucidated. Future preventive/therapeutic strategies for CAA-related hemorrhages should include protection of vessel walls against these factors, as well as the development of anti-amyloid therapies for CAA.
Acknowledgements
The study was supported in part by a grant from the Amyloidosis Research Committee from the Ministry of Health, Labor and Welfare, Japan, and by the SENSHIN Medical Research Foundation, Japan. The author is grateful to Ms. Etsuko Tsujiguchi for her excellent secretarial work.
Notes
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.