Introduction
Although brain atrophy, defined as loss of brain volume unrelated to a defined macroscopic injury [1], is a prominent feature of neurodegenerative disease, it can also reflect cerebrovascular disease. Reduced overall brain volume [2] as well as specific reductions in white matter volume (WMV) [3] or cortical thickness [4] have been observed in various small vessel diseases, often correlating with measures of cognitive impairment. Atrophy of the basal ganglia, however, has generally not been analyzed in the setting of cerebral small vessel disease.
Cerebrovascular β-amyloid deposition or cerebral amyloid angiopathy (CAA) is a common pathology of the leptomeningeal and cortical small vessels that has been linked to cortical hemorrhagic lesions as well as non-hemorrhagic cortical injury such as brain atrophy. Analysis of patients diagnosed with sporadic CAA by validated clinical criteria or with hereditary CAA by genetic testing has found thinning of the cerebral cortex in both groups [5]. Despite its localization to leptomeningeal and cortical small vessels, CAA also appears to cause substantial non-hemorrhagic damage to subcortical structures, including increased white matter hyperintensities of presumed vascular origin [6], lower supratentorial WMVs [7], changes in white matter diffusion-tensor imaging (DTI) properties [8,9], and white matter microinfarcts [10,11]. The likeliest mechanism for CAA-related non-hemorrhagic brain injury appears to be vascular dysfunction [12,13] leading to tissue ischemia. Although few studies have searched for effects of CAA on subcortical gray matter structures such as the basal ganglia, an early DTI study of this disorder reported increased fractional anisotropy (FA) of subthalamic gray matter [8].
Based on the association of CAA with volume loss in cortex and white matter, we sought to determine its relationship to basal ganglia volume (BGV). The basal ganglia, typically defined as putamen, caudate, globus pallidus, and nucleus accumbens, are a set of interconnected deep gray matter nuclei thought to regulate a range of complex functions including voluntary and involuntary motor control [14], non-declarative memory and habit formation [15], language processing [16], action selection [17], and emotional perception [18]. Basal ganglia damage is a prominent early manifestation of neurodegenerative disorders such as Parkinson's disease [19], progressive supranuclear palsy [20], and Huntington's disease [21], where it is felt to mediate neurocognitive as well as movement-related clinical symptoms [22]. From a clinical standpoint, the current finding of CAA-related loss of BGV could represent a novel mechanism for mediating a portion of the cognitive and motor impairments related to this common small vessel pathology and thus an imaging marker for assessing CAA’s impact and its response to candidate interventions.
Methods
Study design and participants
Research cohorts
Individuals (n=80) with sporadic CAA were prospectively enrolled at Massachusetts General Hospital, in Boston, MA, between March 15, 2006 and December 1, 2014. Each individual was non-demented and carried a diagnosis of probable CAA per the pathologically validated modified Boston criteria [23]. Demographic and clinical data were collected for each patient at the time of enrollment.
Of the 80 research patients with sporadic CAA, 68 agreed to undergo a standard neurocognitive battery. Executive function (Trail Making test B, Digit Span test backwards, verbal fluency test), processing speed (Trail Making Test A, Symbol Substitution test), verbal memory (immediate and delayed memory scores based on Hopkins Verbal Learning test), and language processing (Animal Naming test, Boston naming test) z-scores were calculated using the mean and standard deviation scores from the entire sporadic CAA cohort, as described [9]. Gait velocity and time were determined using the Timed Get Up and Go test (n=62) [9].
Individuals in the research CAA cohort were age-matched to healthy controls (HC; n=80) and patients with Alzheimer's disease (AD; n=80) collected from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) [24]. Of the 80 AD patients collected from ADNI, 46 (57.5%) had a clinical dementia rating score of 0.5; the remaining 34 had a clinical dementia rating score of 1.
Individuals with the Dutch-type hereditary form of CAA (D-CAA, previously known as Hereditary Cerebral Hemorrhage With Amyloidosis—Dutch or HCHWA-D) were prospectively enrolled at the Leiden University Medical Center and the diagnosis confirmed by genetic testing [5,25]. The D-CAA patients (n=25) were age-matched to genetically verified mutationnegative HCs (n=25) enrolled at the same center. Of the 25 enrolled D-CAA patients, 14 presented clinically with lobar intracerebral hemorrhage (ICH) and 10 were presymptomatic carriers of the D-CAA-associated gene mutation. Demographic and clinical data were similarly collected for the D-CAA patients [25].
Cohorts with clinical grade magnetic resonance images
In addition to the above cohorts that received high-resolution dedicated research magnetic resonance images (MRIs), we analyzed clinical MRIs of patients who were admitted to Massachusetts General Hospital between January, 2003 and February, 2012, with spontaneous symptomatic ICH. Patients in this group were categorized as having definite or probable CAA-related lobar ICH (CAA-ICH: n=93), and age-matched patients with hypertensive arteriopathy and ICH (HTN-ICH: n=93). The latter group consisted of patients with ICH in the basal ganglia, thalamus, or brainstem but not lobar ICH, as described [26]. All analyses were performed on non-ICH containing hemispheres; patients with bilateral ICHs were excluded. Demographic and clinical information was obtained for those patients at the time of enrollment [26].
Age-matching was performed based on the closest first decimal of age without knowledge of clinical or radiological information [5]. The Institutional Review Boards of the participating sites approved this study and all patients provided signed written informed consent.
MRI acquisition
The research cohort of sporadic CAA patients underwent structural MRI using a Siemens Avanto 1.5T scanner (12-channel head coil; Siemens Healthineers, Erlangen, Germany). The standardized battery included T1-weighted Multi-echo Magnetization- Prepared Rapid Gradient Echo (voxel size 1×1×1 mm3; repetition time [TR], 2,730 ms; inversion time [TI], 1,100 ms), high-resolution susceptibility weighted imaging (SWI; voxel size 0.75×0.75×1.3 mm3; TR, 48 ms; echo time [TE], 40 ms), three-dimensional fluid attenuated inversion recovery (FLAIR; voxel size 1×1×1 mm3; TR, 6,000 ms; TE, 302 ms; TI, 2,200 ms), and high-resolution Diffusion (voxel size 2×2×2 mm3; TR, 8,180 ms; TE, 82 ms; 60 directions; b-value=700; 10 b0 images) sequences.
HCs and patients with AD selected from the ADNI database underwent a similar structural MRI protocol using Siemens 1.5T scanners [5,24].
Patients diagnosed with D-CAA and their matched controls underwent research structural MRI using a Philips 3T scanner (Philips Healthcare, Andover, MA, USA). The MRI protocol included 3D T1-weighted (voxel size 0.875×0.875×1.2 mm3; TR, 9 ms; TE, 4.6 ms), SWI (voxel size 0.7812×0.7812×0.8 mm3; TR, 45 ms; TE, 31 ms), and FLAIR (voxel size 0.4297×0.4297×5.5 mm3; TR, 11 ms; TE, 125 ms) sequences.
The clinical cohorts of patients with CAA-ICH or HTN-ICH underwent a structural MRI protocol using a General Electric Sigma 1.5T scanner (GE Healthcare, Barrington, IL, USA). The clinical MRI protocols included T1-weighted, T2*-weighted Gradient-Recalled Echo, and FLAIR sequences, as described [26].
MRI processing
Structural analyses
Segmentation of cortical gray, white, and basal ganglia structures as well as estimated total intracranial volume (eTIV) were performed using FreeSurfer version 5.3.0 (https://surfer.nmr.mgh.harvard.edu). Cortical gray matter volume (CGMV) was defined as the volume between the pial surface and gray-white junction, measured using a surface-based approach [27] and expressed as a percent of intracranial volume (pCGMV). Similarly, WMV was defined as the volume enclosed within the gray-white junction minus any non-white matter entities such as the subcortical gray matter structures and the ventricles and expressed as a percent of intracranial volume (pWMV). The basal ganglia volume was defined as the sum of the putamen, caudate nucleus, globus pallidus, and nucleus accumbens volumes, and expressed as a percent of intracranial volume (pBGV). White matter hyperintensity volume, a marker of CAA-related structural injury, was estimated using a previously validated semi-automated algorithm, and expressed as a percentage of eTIV (pWMH) [6].
Diffusion analyses
High-resolution diffusion scans were preprocessed using the ‘dt_recon’ pipeline of FreeSurfer. Preprocessing of the scans consisted of correcting eddy currents and head motion artifacts while registering all diffusion scans to a common anatomical template. After manually confirming accurate co-registration between the diffusion and anatomical scans, the resulting FA and apparent diffusion coefficient (ADC) volumetric heat maps were used to calculate the FA and ADC values within the putamen, caudate nucleus, globus pallidus, and nucleus accumbens. Basal ganglia FA and ADC values were defined as the volume-weighted average FA and ADC of those sub-structures.
All segmentation outputs and results were manually checked to ensure accuracy and manual interventions were performed when necessary [5]. Whenever gross pathology (i.e., ICH) was present in either hemisphere (n=40), results were obtained only from the contralateral hemisphere.
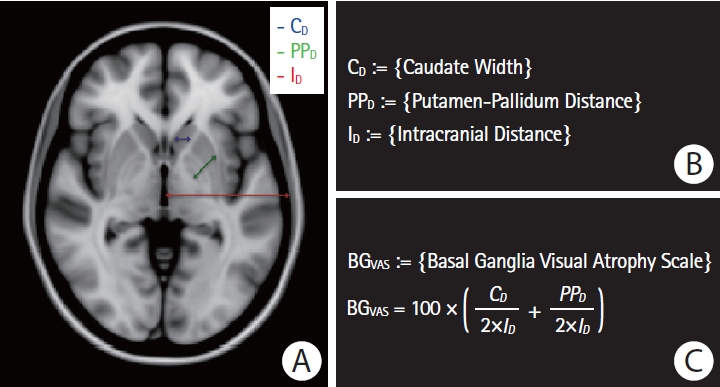
Normalized basal ganglia width
To assess basal ganglia atrophy in ICH patients with clinical- grade MRI scans not suitable for FreeSurfer-based volumetric measurement, we developed a metric based on the sum of the widths of three basal ganglia substructures—the caudate nucleus, putamen, and globus pallidus—normalized to the intracranial diameter (Figure 1). The measurement was obtained from the axial slice containing the largest area of the putamen, anterior commissure, septum pellucidum, and the pulvinar of thalamus [28] in the ICH-free hemisphere of each subject’s axial T1-weighted sequence. An experienced rater (P.F.) determined normalized basal ganglia width on the 93 CAA-ICH and 93 HTN-ICH clinical cases of interest. For comparison to HCs, we applied the same basal ganglia width metric to MRI scans from 93 HC obtained from ADNI (Table 1). To test the metric’s reliability, a second rater (M.P.) independently measured 30 randomly selected CAA-ICH cases and 30 randomly selected HTN-ICH cases. For these 60 scans, the intraclass correlation coefficient (two-way random effects model, consistency, single measures; SPSS software suite version 25, IBM Co., Armonk, NY, USA) was 0.847 (95% confidence interval [CI], 0.756 to 0.906). To assess the validity of the developed basal ganglia visual atrophy scale to accurately estimate BGV, we applied it on the research-quality ADNI HC cohort (n=93) and compared it to their corresponding FreeSurfer-derived pBGV. The Pearson's correlation between the visual atrophy scale and the automated volumetric measurements was r=0.22 (P=0.037).
Statistical analyses
Statistical analyses were performed using the JMP Statistical Software by SAS version Pro 14.0 (SAS Institute Inc., Cary, NC, USA). Bivariate correlation analyses were carried out in the form of two-tailed t-tests (threshold for significance, P<0.05), and Pearson’s correlation coefficients (r). The 240 research subjects (80 sporadic CAA patients, 80 matched HC, and 80 matched AD patients) were included in a general linear model with pBGV as the dependent variable, the binary diagnostic status (“CAA” vs. “HC” vs. “AD”) as the class variable, and other potential confounders (age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus) as covariates. Tukey’s test was used to correct for multiple comparisons between the three groups. Multiple linear regression models were additionally implemented to examine the independent relationship between the dependent variable pBGV and other variables of interest, after adjusting for the potentially confounding effects of age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus. Standardized regression coefficients (β), 95% CI, and P-values are reported for each multiple regression model. All volumetric variables examined were normally distributed.
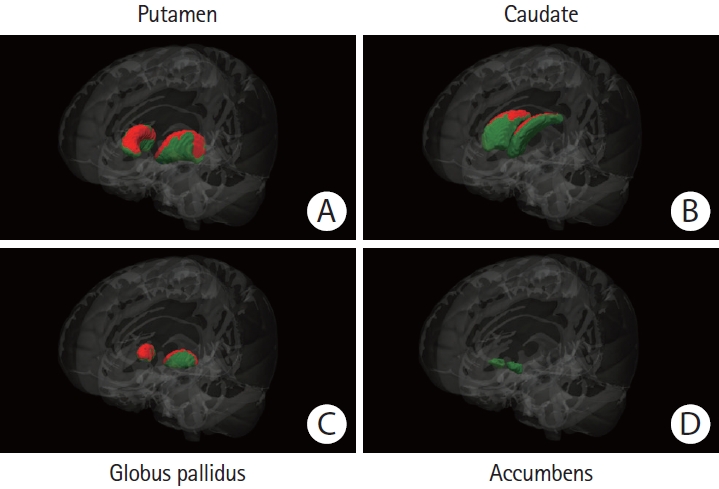
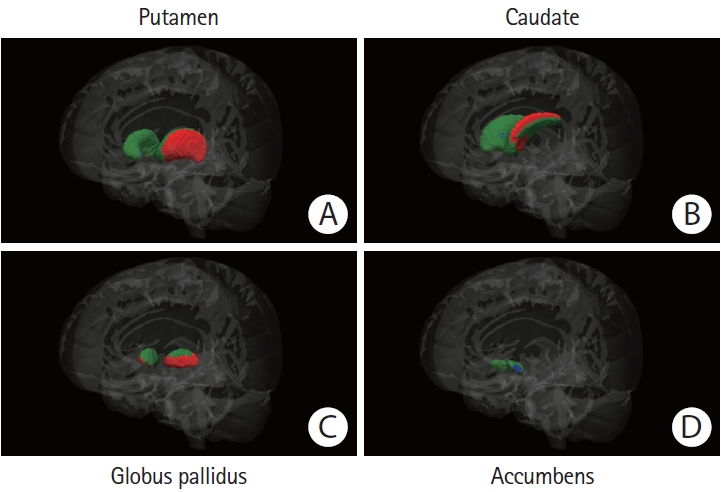
A separate Bayesian appearance model was implemented using Functional Magnetic Resonance Imaging of the Brain (FMRIB) Software Library (FSL) FMRIB’s Integrated Registration and Segmentation Tool (FIRST) automated toolbox in order to schematically explore the regional differences in volume between the basal ganglia and their individual components in patients with CAA, HC, and patients with AD. Topographical maps were generated using a threshold of P<0.05, corrected for multiple comparisons.
Results
Decreased basal ganglia volume in CAA
The research cohort consisted of 80 non-demented patients with sporadic CAA (mean age, 70.3±8.5 years), 80 age-matched HC (71.8±3.7), and 80 age-matched patients with AD (70.4±6.8). Detailed demographic and clinical information for these cohorts are provided in Table 2.
Sporadic CAA patients had lower pBGV than both HC (10.7% lower, P<0.0001) and patients with AD (5.4% lower, P=0.001). Atrophy was identified in the putamen, caudate nucleus, and globus pallidus (Table 3, Figures 2 and 3). Reduced pBGV in CAA relative to HC (pairwise difference=-0.13; 95% CI, -0.18 to -0.09; P<0.0001) and AD (pairwise difference=-0.05; 95% CI, -0.10 to -0.01; P=0.016) remained independent after controlling for age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus, using a general linear model adjusting for multiple comparisons (Tukey’s). Other variables in this model correlating with decreased pBGV were male sex (pairwise difference=-0.10; 95% CI, -0.13 to -0.07; P<0.0001) and presence of hypertension (pairwise difference=-0.05; 95% CI, -0.08 to -0.01; P=0.005).
In a secondary analysis of basal ganglia atrophy relative to total brain atrophy, BGV was expressed as a percentage of total brain volume (defined as the sum of cortical gray matter and WMVs) and compared between the three cohorts. In bivariate analyses, patients with CAA had lower BGV expressed as a perpercentage of total brain volume compared to both HC (4.1% lower, P=0.015) and patients with AD (3.8% lower, P=0.024). The association between BGV expressed as a percentage of total brain volume and diagnostic group did not remain significant at the P<0.05 threshold (CAA vs. HC pairwise difference=- 0.08; 95% CI, -0.17 to 0.01; P=0.08) (CAA vs. AD pairwise difference=-0.07; 95% CI, -0.15 to 0.02; P=0.17) after correcting for age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus (general linear model adjusting for multiple comparisons - Tukey’s).
To further exclude confounding due to the presence of ICH or hypertension, we repeated these analyses in the subgroup of 40 CAA patients (mean age, 71.3±8.1) without ICH versus their corresponding age-matched HC and AD patients, and the subgroup of 34 non-hypertensive patients with sporadic CAA (67.9±8.3) versus their corresponding non-hypertensive agematched HC and AD patients. In both subgroup analyses, patients with sporadic CAA had lower pBGV versus their comparator subgroups (Table 3).
We performed similar analyses of BGV in patients with D-CAA (n=25; mean age, 46.0±14.2) and their corresponding age-matched HC (n=25; mean age, 46.0±11.5). D-CAA patients demonstrated lower pBGV than their matched HC (decrease by 7.2%, P=0.036) (Table 3). This association was again independent of age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus (pairwise difference=-0.09; 95% CI, -0.17 to -0.01; P=0.029). In the same model, increased age was also independently correlated with smaller pBGV (β=-0.43; 95% CI, -0.62 to -0.14; P=0.003). Decreased pBGV in the D-CAA individuals was localized to the putamen (P=0.030) and the nucleus accumbens (P=0.049) with trend towards smaller globus pallidus (P=0.083) but not caudate nucleus (P=0.42).
To analyze for basal ganglia atrophy using clinical (rather than research-quality) MRI scans from patients with CAA-ICH and HTN-ICH, we developed a reproducible technique (see Methods) for measuring the normalized width of the caudate, putamen, and globus pallidus. Patients with CAA-ICH (n=93; mean age, 68.5±7.1) were compared to similar aged patients with HTN-ICH (n=93; 68.5±12.3) and an ADNI HC cohort (n=93; 73.1±3.3) (Table 1). Patients with CAA-ICH had greater basal ganglia atrophy versus HTN-ICH (8.3% decrease in normalized basal ganglia width, P<0.0001) and both ICH groups had greater basal ganglia atrophy versus HC (CAA-ICH vs. HC: 20.1% decrease, HTN-ICH vs. HC: 12.9% decrease, P<0.0001 for both comparisons) (Table 1). Using a general linear model adjusting for multiple comparisons, the association between CAA-ICH and greater basal ganglia atrophy was again independent after correcting for age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus, as well as history of prior ICH and ischemic stroke (CAA-ICH vs. HTN-ICH pairwise difference=- 1.11; 95% CI, -1.61 to -0.62 and CAA-ICH vs. HC pairwise difference=-3.18; 95% CI, -3.82 to -2.53; P<0.0001 for both comparisons). In the same model, male sex was independently associated with decreased BGV (pairwise difference= 0.84; 95% CI, 0.52 to 1.17; P<0.0001).
Basal ganglia volume and other markers of structural integrity
We previously reported CAA-related cortical and white matter atrophy [5,7] as well as elevations in subcortical gray matter FA [8] and explored the association of these parameters with CAA-related basal ganglia atrophy. Among sporadic CAA patients, reduced pBGV correlated with pCGMV (r=0.45, P<0.0001) but not pWMV (r=0.18, P=0.12) or pWMH (r=0.02, P=0.85). The positive correlation between pBGV and pCGMV remained independent after adjusting for age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus (β=0.33; 95% CI, 0.05 to 0.65; P=0.011). The same subjects demonstrated a negative correlation between pBGV and basal ganglia FA (r=-0.36, P=0.001), and no association between pBGV and basal ganglia ADC (r=0.05, P=0.68). The correlation between pBGV and basal ganglia FA was also independent of age, sex, presence of hypertension, hyperlipidemia, and diabetes mellitus (β=-0.24; 95% CI, -0.45 to 0.03; P=0.031).
Basal ganglia volume and neurologic function
Because of the association of CAA with cognitive impairment [29], we performed an exploratory analysis of the relationship between BGV and cognitive performance. Among the research cohort sporadic CAA patients who underwent complete neurocognitive assessment (n=68), there was a positive correlation between pBGV and verbal memory (r=0.26, P=0.034) and language processing (r=0.35, P=0.003), but not with executive function (r=-0.05, P=0.67) or processing speed (r=-0.13, P=0.30). The positive association between pBGV and language processing was independent of age, sex, presence of hypertension, hyperlipidemia, diabetes mellitus, years of education, as well as pCGMV and pWMV (β=0.35; 95% CI, 0.04 to 0.66; P=0.013) whereas the association with verbal memory did not persist in this model (β=0.13; 95% CI, -0.23 to 0.44; P=0.44). There was no association between pBGV and gait velocity (r=0.02, P=0.91) or gait time (r=-0.13, P=0.30).
Discussion
Our primary finding is that CAA is associated with reduced BGV. Total BGV was reduced by 10.7% and 7.2% respectively in non-demented sporadic and hereditary CAA patients relative to age-matched HCs, with similar decreases observed in the individual volumes of putamen, caudate, and globus pallidus. BGV as a whole and the individual putamen, caudate, and globus pallidus volumes were also reduced in CAA relative to AD patients, while nucleus accumbens was smaller in AD than CAA, consistent with the high burden of AD pathology in this structure [30]. Measuring the normalized widths of caudate, putamen, and globus pallidus, we found basal ganglia atrophy in both CAA-ICH and HTN-ICH relative to HCs, but to a greater extent in CAA-ICH than HTN-ICH.
Effects of CAA on BGV are somewhat unexpected given CAA’s primarily cortical and leptomeningeal localization; the current findings should therefore be considered exploratory and requiring of replication. CAA-related effects on subcortical brain regions such as the white matter, however, are well established. Amyloid positron emission tomography imaging, for example, showed a direct relationship between increasing brain amyloid burden and volume of white matter hyperintensity in non-demented CAA patients but not AD patients [6]. The mechanisms for these distant effects on subcortical brain regions remain undetermined, but could include (1) subcortical ischemia due to cortical vascular dysfunction and distal hypoperfusion and (2) subcortical neurodegeneration due to loss of cortical afferent and efferent connections. Indeed, our primary and secondary analyses together indicate that basal ganglia atrophy in CAA is to some extent in proportion to total brain volume loss, which might result either from loss of afferent and efferent connections across compartments or from correlations in the extent of injury among the brain compartments driven by overall CAA severity in each brain. An early postmortem study of eight brains with CAA noted demyelination, vacuolation, and lacunae in white matter and basal ganglia in a pattern similar to Binswanger’s subcortical encephalopathy [31], while another postmortem study of 18 brains with severe CAA found frequent microinfarcts in the white matter as well as cortical gray matter [10]. Moreover, CAA-ICH patients have previously been reported to have a similar burden of WMH as HTN-ICH [32] and in the current study demonstrate greater basal ganglia atrophy. These findings suggest that the relatively distant effects of CAA on subcortical brain structures equal or exceed those caused by arteriolosclerosis and lipohyalinosis of the more proximate deep penetrating arterioles in HTN-ICH [33].
We found that reduced BGV among the sporadic CAA research cohort correlated with higher basal ganglia FA. DTIbased metrics such as FA are typically studied in white matter, where higher FA is generally interpreted as indicating greater directionality of water diffusion along myelinated tracts [34]. The direction of the currently reported relationship suggests that higher FA correlates with greater basal ganglia injury, analogous to an earlier DTI study in CAA demonstrating increased FA in subthalamic gray matter relative to HCs [8]. These data raise the possibility that when measured in subcortical gray matter nuclei, increased FA may indicate greater structural injury. Indeed, this idea has also been suggested in studies of striatum and pallidum in early Huntington’s disease [35] and caudate in relapsing- remitting multiple sclerosis [36]. A postulated mechanism for increased subcortical gray FA is targeted loss of dendritic connections causing restricted directional diffusion of the underlying fiber tracts [36].
It remains to be determined whether CAA-related basal ganglia atrophy is a major contributor to the disease’s clinical manifestations. Although we found relatively strong correlations between the extent of atrophy and two cognitive domains, only the association with language processing remained independent after controlling for covariates. This is noteworthy, especially considering how other types of CAA-related structural atrophy, such as white matter atrophy, affect different domains of cognitive function [7]. It could thus be speculated that basal ganglia atrophy in patients with CAA is driven by a different underlying pathological mechanism and could contribute to the disease’s long-term neurocognitive dysfunction. Previous studies in neurodegenerative diseases have indeed pointed to a possible role for basal ganglia atrophy in cognitive impairment [22,37]. Given the potential for confounding by other CAA-related brain injuries [38], however, it remains unclear whether the correlation observed among the research CAA patients indicates direct functional impact of basal ganglia atrophy.
In addition to difficulty identifying the independent contribution of basal ganglia atrophy to CAA-related clinical dysfunction, the current analysis has other limitations. Even nondemented CAA patients typically have some degree of concomitant AD pathology, raising the possibility that the identified basal ganglia atrophy is due to AD rather than CAA. We found lower caudate, putamen, globus pallidus, and total BGVs in CAA relative to AD, however, arguing against this interpretation. Similarly, basal ganglia atrophy in D-CAA should be largely unconfounded by AD pathology, as this condition has relatively few mature senile plaques and minimal neurofibrillary pathology [39]. Another limitation is the use of controls and AD scans drawn from ADNI for comparison to our CAA research and clinical cohorts, introducing the possibility of spurious differences due to variations in the MRI scans. We note that the ADNI patients were imaged using the same scanner type and magnetic field strength as the CAA group, and that the comparisons yielded basal ganglia regions that were both smaller in CAA than AD (caudate, putamen, globus pallidus) and smaller in AD than CAA (nucleus accumbens) suggesting no consistent inter-scanner bias. We also note that D-CAA patients and their matched controls were scanned on the same scanner and thus not affected by inter-scanner differences. Our analyses of HTN-ICH required a visual method for determining normalized basal ganglia width that could be applied to clinical-grade scans, potentially introducing inaccuracies in measurement relative to volumetric methods. The significant but not extremely strong correlation between the basal ganglia visual atrophy metric and the FreeSurfer-derived BGVs applied on the comparative HC cohort (r=0.22) suggests that the visual atrophy scale can be used as a basal ganglia atrophy measure when volumetric measurement is not possible, but likely introduces measurement error into the comparison of the CAA-ICH and HTN-ICH patients. The measurement error, however, would be expected to bias towards a null result rather than towards a difference between the groups. The high inter-rater reliability of the visual scale suggests that the method is reproducible and can be tested in future cohorts. Finally, all our analyses were cross-sectional using MRIs obtained at single timepoints. Longitudinal analysis of BGV will be an important future step for characterizing this process.
If confirmed in replication studies, the finding of basal ganglia atrophy would substantially extend the range of subcortical regions affected by CAA, raising challenging questions about the mechanisms for tissue injury distant from the amyloid- laden leptomeningeal and cortical vessels. Basal ganglia atrophy might also represent a novel neuroimaging marker of CAA-related brain injury. Previous studies have identified a range of candidate outcome markers for CAA, including both hemorrhagic and non-hemorrhagic brain lesions as well as alterations in vascular physiology [40]. If future studies suggest that basal ganglia injury indeed contributes to the clinical manifestations characteristic of CAA, then this marker might play an important role in interventional trials aimed at limiting CAA-related neurologic dysfunction.
Conclusions
In this case-control study, patients with sporadic or hereditary CAA displayed significant atrophy in the basal ganglia compared to similar aged individuals without CAA. Moreover, CAA-related basal ganglia atrophy independently correlated with cortical gray matter atrophy, increased basal ganglia fractional anisotropy, and worse performance on language processing. Overall, the independent association of CAA with basal ganglia tissue loss could suggest a new mechanism for CAA-related brain injury and neurologic dysfunction, and a novel candidate biomarker of this currently untreatable disorder.