Introduction
Vascular dementia (VaD) is caused by cerebrovascular disease that directly or indirectly damages the brain structures associated with cognitive functioning. Historically, the concept of VaD has been described since the 19th century when arteriosclerotic brain atrophy due to hardening of the arteries was perceived as the major cause of senile dementia.1,2 The importance of VaD was overshadowed in the past century by the emergence of Alzheimer's disease (AD), which has turned out to be the most common cause of dementia. Interest in VaD has been revived in recent years because vascular lesions have been continuously noted to be substantial contributors to the development of dementia by themselves or by their synergetic effects on the pathogenesis associated with AD.3,4 With accumulating evidences suggesting the role of vascular burden on cognitive functions, VaD has now evolved into the concept of vascular cognitive impairment (VCI), which encompasses not only VaD but also AD with cerebrovascular disorder (AD with CVD or mixed dementia) and VCI with no dementia (VCIND). Thus, VCI is a more broad term that comprises all the states of cognitive impairment associated with CVD.3-5 Around one-third of patients with AD demonstrate evidences of vascular pathology,6-8 while up to two-thirds of patients with CVD develop at least some degree of AD pathology in the brain.9,10 Subcortical ischemic vascular dementia (SIVD) is one of the VaDs which can have both vascular and AD associated pathology in the brain. SIVD is pathologically driven by stenosis and occlusion of small vessels, resulting in white matter ischemia and multiple lacunar infarctions in subcortical structures. SIVD is of particular interest as the relatively slow progression of symptoms and clinical manifestations of the disease often make the differentiation of it from AD difficult. In this article, the authors reviewed the emerging concepts of VaD/VCI and clinical manifestations, biomarkers, treatments, and preclinical models of SIVD based on the pathophysiologic mechanisms of the disease.
Emerging concepts in VaD and VCI
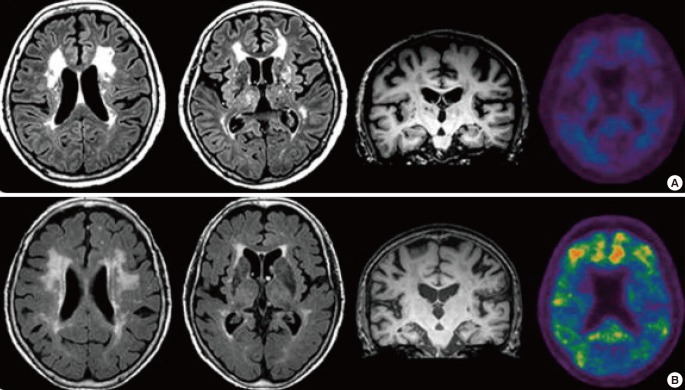
VaD can be classified into the following: 1) multi-infarction dementia, 2) strategic infraction dementia, 3) hemorrhagic dementia, 4) mixed dementia, 5) SIVD, and 6) other forms of vascular dementia.5,11 The first three types of VaDs develop relatively suddenly because of acute cerebrovascular diseases and they demonstrate specific cortical or subcortical symptoms depending on the stroke-affected regions. The stepwise progression, fluctuation of symptoms, and focal neurological signs are suggestive of acutely developing VaD. However, the cognitive impairments associated with SIVD often show an insidious onset and progressive decline; thus, mimicking the course of AD.12 Motor symptoms are often not obvious and go unnoticed in this setting, further confusing clinicians. It is suggested that the AD pathologies are the leading cause of dementia and this can take precedence over the status of cerebral ischemia.12-15 A recent amyloid imaging study, however, has indicated that there exists a pure SIVD, in which significant subcortical white matter ischemic changes are present with no evidence of amyloid plaque deposition in the brain (Figure 1).16 Moreover, pure SIVD is more prevalent than previously thought. This condition is distinctive from AD or mixed dementia, which demonstrate fibrillar forms of amyloid deposition in the brain.16,17 Finally, other forms of VaD include dementia that is caused by heterogeneous etiologies (e.g., vasculitis, cerebral amyloid angiopathy, and hereditary diseases, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy [CADASIL]).18-20
There is growing consensus on the concept of mild cognitive impairment (MCI) due to AD or very mild AD. It is now believed to be early phases of the pernicious accumulation of amyloid beta (Aβ) plaques and neurofibrillary tangles in the brain, with diverse ranges of severity in clinical symptoms.21,22 In contrast, the concept of VCI emphasizes the treatable and preventable components of dementia, including the vascular burden in the brain. As such, the concept of VCI has evolved to amplify the potential treatment of vascular factors in order to improve the cognitive impairments because some vascular components are modifiable to prevent additional cerebrovascular deterioration and to ameliorate the current or ongoing cognitive impairments. Even after cognitive impairment sets in, the modulation of vascular risk factors could provide secondary prevention and improvement of the cognitive deficits.5,11,13
In line with the emerging concept of the VCI, cerebral small-vessel disease has drawn lots of interest in the field of AD. The prevalence and influence of cerebral small-vessel disease are considered to be significant in dementia, although underestimated so far.23 Evidence of small vessel disease found on magnetic resonance images (MRI; e.g., white matter hyperintensities or lacunar infarctions) can be observed in 20%-40% of community-dwelling older persons12 and the prevalence of cognitive impairment in association with small vessel disease has been estimated to be as high as 36%-67%.13,24 In addition, recent studies represent that AD also increased the incidence of stroke.14,15 Thus, it is worthwhile to compare SIVD to AD in terms of the pathophysiological mechanisms, clinical manifestations, biomarkers, treatment options, and preclinical models.
Pathophysiology of SIVD
SIVD is mainly caused by widespread ischemic changes in the white matter or multiple lacunar infarctions in subcortical structures.13,23,25-28 Predominant arteriosclerotic leukoencephalopathy in the white matter is classically referred to as Binswanger's disease. This type of ischemic change in the brain is formed by widespread incomplete infarctions or hypoperfusion of the white matter due to critical stenosis in the cortical medullary branches. Pathologically, the white matter lesions demonstrate degeneration of the myelin sheath and axons and degradation of the oligodendrocytes without profound infarction or cystic necrosis. On the other hand, the lacunar infarctions (état lacunaire) are caused by perforating arteriolar occlusions in subcortical structures, including the thalamus, basal ganglia, and internal and external capsules. The possible mechanisms underlying the steno-occlusion of small vessels include increased resistance to blood flow, decreased autoregulation, disruption of the blood-brain barrier (BBB), dysfunction of the endothelium, and dilatation of the perivascular spaces.12,23,27 The ultimate disruption of the prefronto-subcortical circuits and thalamocortical circuits by the accumulation of white matter ischemic lesions and lacunar infractions is thought to be an underlying mechanism that result in the cognitive impairments in SIVD. In mixed dementia, which has both the vascular and AD pathologies in the brain, these alterations in cerebrovascular circulation have been described to contribute to the development of dementia by themselves or by a synergistic interaction with AD pathology in the brain.29 However, the concomitant pathology of AD, including cholinergic deficits and the pathologic accumulations of Aβ plaques and neurofibrillary tangles, has also been found in patients with alleged SIVD, which is also believed to accelerate cerebral ischemia in the brain.16
Clinical manifestations of SIVD and its associations with structural and functional brain images
Along with cognitive impairments, the stepwise development of neurological deficits is not uncommon in patients with SIVD. These deficits include hemiparesis, dysphagia, dysarthria, pseudobulbar palsy, emotional incontinence, urinary incontinence, and parkinsonian features, such as short-stepped gait. Abnormal behaviors, including disinhibition and akinetic mutism, can also develop during the course of SIVD, if the prefronto-subcortical circuits are interrupted directly or indirectly.30 Because behavioral changes usually develop later in the course of AD, the early appearance of behavioral or psychological symptoms can suggest the existence of SIVD, particularly when they present in stepwise patterns. Neuropsychological tests can reveal impairments in attention, execution, set-shifting performances, and verbal fluency from the early phases of SIVD. However, patients with AD demonstrate more difficulties with memory, naming, and visuospatial functions from the beginning of the symptomatic phases. A recent amyloid positron emission tomography (PET) imaging study has shown that patients with AD having fibrillar forms of Aβ in the brain demonstrate more difficulties in verbal/visual memory-associated tasks, while those with pure SIVD without amyloid pathology in the brain exhibit more trouble during executive functions, such as phonemic fluency of the Controlled Oral Word Association Test and the Stroop color test (Table 1).17,31-33 Associations between the amount of white matter pathology and impairments in daily living activities have also been described.34
Beyond the signal changes visible on MRI, recent methodological advances have enabled further image-based analyses to investigate the integrity of normal-appearing white matter tracts and the mechanisms that underlie cognitive dysfunctions associated with white matter changes. Previous studies on patients with CADASIL described an increased mean diffusivity and decreased diffusion anisotropy in the areas of white matter hyperintensities, which suggests the disruption of the white matter tracts in SIVD.35,36 Although the interpretation is limited due to the small numbers of participants, previous studies on patients with SIVD reported perfusion defects in the bilateral pulvinar nuclei of the thalamus,37 caudate nucleus, and diverse cortical areas, including the cingulate, superior temporal, subcallosal gyri,38 and frontal lobes.39 These findings support the notion of frontosubcortical circuit disruption in patients with SIVD. In addition to structural imaging biomarkers, PET images obtained with amyloid tracers can also be very useful for demonstrating the underlying pathology of SIVD. Although recent guidelines from the Amyloid Imaging Task Force convened by the Alzheimer's Association and the Society of Nuclear Medicine and Molecular Imaging do not recommend using amyloid PET imaging as a routine test for the diagnosis of AD,40,41 amyloid PET imaging can be clinically useful for differentiating pure SIVD and mixed dementia, which would sometime be tough to distinguish and would pose diagnostic dilemmas. A new criterion that could help discriminate pure SIVD from mixed dementia is under way using a combination of clinical and MRI findings based on data from amyloid PET imaging.
Preclinical models of SIVD
Two different models of SIVD have been described in the literature. One is a mechanical model, in which microcoils are applied to the bilateral common carotid arteries to induce continuous hypoperfusion and ischemia in the white matter without apparent gray matter changes.42,43 This mouse model has demonstrated decreased hippocampal volume and impaired working memory deficits that are possibly due to damaged frontosubcortical circuits. Subsequent demyelination of the white matter tracks is typical in this model, which can mimic the demyelinating pathology of the SIVD. However, the lack of methods that can affect the actual small vessels is a fundamental limitation of this model, even though the manipulation of the carotid artery does mimic the pathological findings of SIVD. The other model of SIVD is a transgenic mouse model of CADASIL that expresses Notch3 mutations at R90C.44 As CADASIL shares many pathophysiological and clinical manifestations with SIVD, research on SIVD and CADASIL is closely associated. Mouse models of CADASIL also develop similar cerebrovascular changes in the brain as in humans.45,46 However, the limited volume of white matter in rodents is a drawback and mouse models of CADASIL do not demonstrate widespread white matter changes in the brain. Recent advances in technology will make the modulation of the small vessels easier and more reliable. If new mouse models of SIVD can be introduced, it will allow better understanding of the direct pathological burden of the CVD on cognition, which can be separated from other cognitive impairments driven by neurodegenerative etiologies, such as accumulation of Aβ plaques or neurofibrillary tangles.
SIVD Biomarkers
Various results have been described in biomarker studies on patients with VaD/VCI. A recent longitudinal study has compared the baseline cerebrospinal fluid (CSF) biomarkers of four different groups: a MCI group that finally progressed to AD (MCI-AD), mixed dementia (MCI-MD), or SIVD (MCI-SIVD), and a MCI group that remained stable as MCI (MCI-MCI).47 The results from CSF Aβ and tau showed that the levels of Aβ were higher and the levels of total tau and the phosphorylated forms of tau (p-tau) were lower in the MCI-SIVD group than in the MCI-AD group. Interestingly, the levels of CSF biomarkers obtained in the MCI-MD group were in between the levels of the MCI-AD group and the MCI-SIVD group. The CSF levels obtained from the MCI-SIVD group were closer to the levels of the MCI-MCI group and the normal control group. The intermediate CSF levels of Aβ and tau in the SIVD group compared to the mixed dementia/AD and normal controls indicate that SIVD possibly shares a common pathophysiology with AD. When patients with VaD and AD are compared, the levels of CSF Aβ had a tendency to be lower and CSF tau had a tendency to be higher in patients with AD than in patients with VaD, but substantial overlaps existed between the groups.48,49 On the other hand, another study that compared the levels of CSF Aβ and tau among non-demented elderly persons who had mild, moderate or severe white matter hyperintensities, demonstrated no differences in the levels of CSF Aβand tau among the groups.50
In addition, other markers associated with BBB breakdown have been introduced as alternative biomarkers associated with SIVD. A compromised BBB can be a source of albumin leakage, which can increase protein concentration in the CSF as it crosses the BBB with less resistance. With this pathological evidence of BBB disruption in patients with SIVD, the increased levels of albumin have been described as supportive evidence of SIVD.51-53 Metalloproteinases are markers of neuroinflammation as they attack the basal lamina and tight junctions in blood vessels, in addition to the disruption of myelin.54 In comparison with AD, the levels of metalloproteinases-9 in the CSF were increased in patients with VCI.55,56 Another marker of white matter disruption is neurofilament, which functions as a cytoskeleton in large myelinated axons.57 The concentrations of the neurofilament light subunit were reported to be higher in subjects with SIVD.47,58 Similar findings were noted in nondemented subjects with severe white matter lesions.50 As briefly described in the previous section, advanced imaging technologies can also serve as biomarkers, which enable further discrimination of the underlying pathophysiologies of SIVD.
Treatment of SIVD
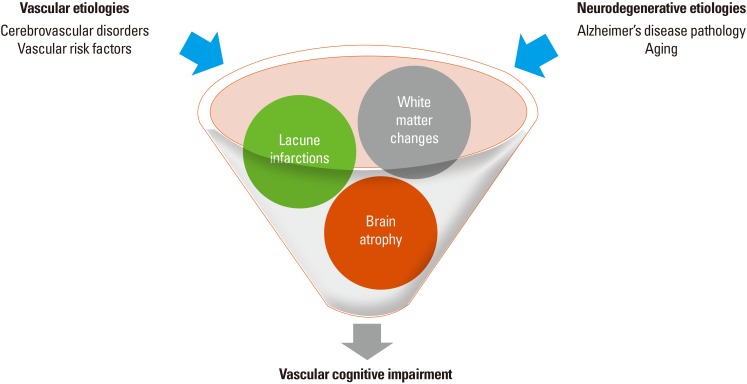
SIVD treatment strategies include both slowing or mitigating underlying small vessel disease progression and the improvement of clinical symptoms. Microinfarction pathology stands out as a significant and independent factor that contributes to brain atrophy and cognitive impairment.26,59,60 Therefore, as a secondary prevention of stroke by modulating small vessel disease, antiplatelet agents can be administered. However, the effectiveness of antiplatelet therapy for the primary prevention of VCI has not yet been established.61 A calcium-channel blocker, nimodipine, has been introduced as a SIVD treatment because of its vasodilation effects. Following potential success in early trials of nimodipine in patients with VaD, an intention-to-treat trial was conducted with a total number of 230 patients. In that study, treatment with nimodipine did not reach the primary endpoint defined by the researchers, although the treatment group demonstrated improved lexical production and better results on the Mini-Mental State Examination and Global Deterioration Scale.62,63 In addition, cholinesterase inhibitors have been introduced to manage cognitive symptoms. Cholinergic deficits in SIVD were described based on findings that the basal forebrain cholinergic nuclei are supplied by perforating arterioles and that the CA1 sector of the hippocampus is vulnerable to ischemic insults.64,65 Patients with CADASIL have also demonstrated deficits in cholinergic fibers. Accordingly, cholinesterase inhibitors have been introduced in the treatment of VaD for the cognitive impairments associated with cholinergic deficits but not so much for other cognitive dysfunctions.66 A recent randomized controlled drug trial on patients with CADASIL has also reported the potential beneficial effects of cholinesterase inhibitors on cognitive functions.67 In another study of CADASIL, treatment with the cholinesterase inhibitor, donepezil, did not improve the primary endpoints measured by the vascular Alzheimer's disease assessment scale-cognitive subscale (VADAS-Cog) scores. However, a subgroup analysis indicated improvements in executive functions in the treatment group.67 Antidepressants, such as selective serotonin reuptake inhibitors, can also be used for the management of depression and atypical neuroleptics can be selectively used for behavioral changes, including agitation and aggression. Recently, modification of cardiovascular risk factors has been recommended for the prevention of AD and VCI, including the SIVD. The modifiable and preventive risk factors include hypertension, diabetes, and hypercholesterolemia. In addition, modifications of lifestyle, including education, diet, physical activity, alcohol consumption, smoking, obesity, and social support/networking, can also be considered for the management of VCI (Figure 2).61
Conclusions
VaD is a heterogeneous disease entity that encompasses various conditions. SIVD is a relatively homogeneous vascular dementia that can mimic AD due to the slow progression of cognitive decline. Detailed medical histories and neuropsychological tests can reveal early impairments in frontal executive symptoms and behavioral or mood changes. Neuroimaging studies and CSF biomarkers can also help in diagnosing and differentiating SIVD from other causes of dementia. Proper and early diagnosis, with careful review of the patients' medical history, neurological findings, structural and/or functional neuroimaging, and information from biomarkers, will enable clinicians to select the proper management techniques for the secondary prevention of stroke and the symptomatic treatment of SIVD. An improved understanding of the pathophysiology of SIVD with diverse experimental models of SIVD as well as the development and validation of new biomarkers will make the diagnosis and treatment of SIVD more reliable and effective. Finally, well-designed longitudinal studies with imaging and biological biomarkers in SIVD and subcortical vascular MCI, such as the Clinical Research Center for Dementia of South Korea (CREDOS),68 the Amyloid PET Imaging for Subcortical Vascular Dementia study (AMPETIS),16 and the Korean Alzheimer's Disease Neuroimaging Initiative (K-ADNI), will shed light on efforts to better understand the pathophysiology and prognosis of SIVD.
Future directions
Data regarding VaD, including SIVD, need to be collected, maintained as a database in a standardized fashion, and shared among researchers to better define and manage dementia. The standardization of vascular lesions in neuropathology and neuroimaging is well under way. More work remains to be done before the concept of SIVD is widely accepted and put actively into clinical practice.