Asymptomatic Cerebral Small Vessel Disease: Insights from Population-Based Studies
Article information
Abstract
Cerebral small vessel disease (CSVD) is a common group of neurological conditions that confer a significant burden of morbidity and mortality worldwide. In most cases, CSVD is only recognized in its advanced stages once its symptomatic sequelae develop. However, its significance in asymptomatic healthy populations remains poorly defined. In population-based studies of presumed healthy elderly individuals, CSVD neuroimaging markers including white matter hyperintensities, lacunes, cerebral microbleeds, enlarged perivascular spaces, cortical superficial siderosis, and cerebral microinfarcts are frequently detected. While the presence of these imaging markers may reflect unique mechanisms at play, there are likely shared pathways underlying CSVD. Herein, we aim to assess the etiology and significance of these individual biomarkers by focusing in asymptomatic populations at an epidemiological level. By primarily examining population-based studies, we explore the risk factors that are involved in the formation and progression of these biomarkers. Through a critical semi-systematic review, we aim to characterize “asymptomatic” CSVD, review screening modalities, and draw associations from observational studies in clinical populations. Lastly, we highlight areas of research (including therapeutic approaches) in which further investigation is needed to better understand asymptomatic CSVD.
Introduction
Several of the recent developments [1-5] in stroke care have focused on the treatment of large vessel occlusions and pathologies. However, there have been few advancements in the management of cerebral small vessel disease (CSVD) largely because small vessels are difficult to observe radiographically and their underlying pathogenic mechanisms are incompletely understood [6,7]. This is problematic given that CSVD contributes to a number of clinically relevant sequelae including hemorrhagic stroke, vascular cognitive impairment (VCI), gait disturbances including parkinsonism, bladder dysfunction, and epilepsy [7-13]. Furthermore, a large proportion of ischemic stroke and dementia are attributed to CSVD (20% and 45%, respectively) [14]. One of the greatest challenges is that there are limited means to assess CSVD prior to the development of its clinical sequelae. Moreover, even after identification of CSVD in healthy populations, management of this disease is not well established. In this review, we aim to summarize basic definitions of CSVD and CSVD biomarkers, highlight our current understanding of CSVD as it relates to “healthy” populations, and where relevant, incorporate evidence from observational studies in different clinical populations. We will focus on asymptomatic CSVD, defined as neuroimaging evidence of CSVD prior to the development of any overt clinical symptoms. Although we will review the major population-based studies in the field, we will crosslink our findings with those of clinical cohorts to unravel clinical relevance and put data into perspective. The emphasis of this paper will be sporadic non-amyloid CSVD (often termed “hypertensive arteriopathy”) [7], as there are several recent reviews that discuss monogenetic CSVD and cerebral amyloid angiopathy (CAA)-related CSVD [15-18].
Methods
Search strategy and selection criteria
Articles from January 1951 to July 2018 were identified through searching the PubMed (National Center for Biotechnology Information, National Library of Medicine) database. The following title/abstract search terms were employed: “cerebral small vessel disease AND population,” “silent lacunar infarcts AND population,” “white matter hyperintensities AND population,” “cerebral microbleeds AND population,” “perivascular spaces AND population,” “cortical superficial siderosis AND population,” and “cerebral microinfarcts AND population.” Exclusion terms were not used in the search criteria. Searches were limited to full-text articles available in English. Additional references were selected by reviewing the reference lists of relevant publications. As this review was restricted to asymptomatic disease, most articles discussing symptomatic stroke or intracerebral hemorrhage (ICH) were excluded. Furthermore, we excluded literature pertaining to CAA given that our focus was on sporadic, non-amyloid CSVD.
Data analysis
By using the above search methodology, 642 articles were generated. Articles were independently screened by two authors (A.S.D. and R.W.R.) for appropriateness and relevance to the topic. Two hundred and eight articles were ultimately selected for incorporation in this review. Using this analysis, we generated a semi-systematic review.
CSVD definitions
CSVD is a broad term that incorporates both neuroimaging and neuropathological findings that pertain to smaller vessels (5 μm to 2 mm) in the gray and white matter including arterioles, capillaries, and venules [6,7,14,19]. While both concepts have been raised in the literature, it should be noted that neuroimaging markers reflect CSVD, but do not define the disease in its entirety. CSVD is often diagnosed by neuroimaging features seen occasionally on computed tomography (CT), but more sensitively detected by magnetic resonance imaging (MRI). These MRI markers include white matter hyperintensities (WMHs) of presumed vascular origin, recent small subcortical infarcts and lacunes, cerebral microbleeds (CMBs), perivascular spaces (PVSs), cortical superficial siderosis (cSS), brain atrophy, and cerebral microinfarcts (CMIs) (Figure 1) [7,14]. In 2013, these imaging markers (with the exception of CMI) were defined by the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) consortium, which unified definitions of CSVD biomarkers based on key neuroimaging characteristics [20]. In most cases, not all of these neuroimaging markers will be present together in a single brain scan. Whether the presence or anatomical location of certain imaging markers reflects distinct small vessel pathways is not fully understood.
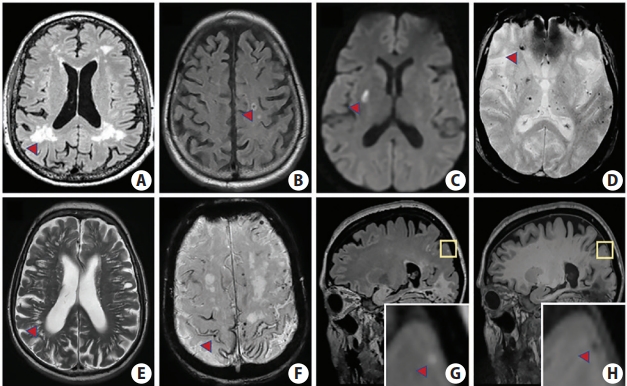
Imaging features of cerebral small vessel disease biomarkers on magnetic resonance imaging. (A) T2 fluid attenuated inversion recovery (FLAIR) sequence depicting white matter hyperintensities (red arrowhead) which are seen as hyperintense regions in the white matter. (B) Lacune (red arrowhead) on FLAIR sequence characterized by a central hypointensity with a surrounding rim of hyperintensity. (C) Recent subcortical infarct (red arrowhead) on diffusion-weighted imaging sequence between 3 to 15 mm in diameter. (D) Susceptibility-weighted imaging (SWI) sequence showing cerebral microbleeds (red arrowhead) which are round/oval shaped signal voids ≤10 mm and have associated blooming artifact. (E) Perivascular spaces (red arrowhead) on T2 which are linear cavitations that do not have a surrounding rim of hyperintensity and are <3 mm in diameter. (F) Cortical superficial siderosis (red arrowhead) visualized on SWI and characterized by a curvilinear hypointensity that follows the gyral surface. (G) Cerebral microinfarct (<5 mm in diameter) is hyperintense on T2 FLAIR (see inset, red arrowhead) and (H) hypointense on T1 (see inset, red arrowhead). Images (G) and (H) were graciously provided by Susanne Van Veluw.
While the true burden of CSVD is not known, estimates suggest that at least one-third of healthy-populations have lacunes (the majority of which are due to CSVD), although the actual prevalence of CSVD is likely much higher [21,22]. While these, often called “silent strokes,” are visible on CT or MRI, they portend no clinical syndrome because of their small size and noneloquent location (sparing motor cortices, cranial nerves, or language centers). However, in patients without CSVD visible on 1.5T MRI, infarcts may still be present, but visible only on ultra-high resolution (7T) MRI images or by pathologic examination. The burden of these CMI can be formidable, with estimates suggesting hundreds to thousands of infarcts in a single brain [23-25]. This fact underscores the difficulty of diagnosing and ascertaining the extent of CSVD, such that the mere presence of one biomarker such as WMH can reflect a much larger process involving several pathological disruptions to the cerebral microvasculature [26].
CSVD is a dynamic process in which lesions including WMH, CMB, and lacunes can progress or regress (even after accounting for differences in MRI technique) [27,28]. The implications of this on CSVD is not well understood but may be a balance between small vessel arteriopathy and neural repair processes. Hypertension and other vascular risk factors are believed to be the primary culprits of this entity as we will see from several epidemiological studies. Herein, we aim to review the epidemiological studies that pertain to each of the major neuroimaging markers. We limit discussion of brain atrophy, given that it can reflect a host of underlying mechanisms beyond CSVD.
White matter hyperintensities of presumed vascular origin
WMH of presumed vascular origin, often termed leukoaraiosis in early reports [29-31], are of variable size, but appear hyperintense on T2-weighted (T2) and fluid attenuated inversion recovery (FLAIR) MRI sequences and hypointense/isointense on T1-weighted sequences [20]. They are more difficult to detect on CT, but may be seen as hypodensities (or hypoattenuated areas). Typically, they spare the subcortical U-fibers, and presumably are due to chronic hypoperfusion with increased blood-brain barrier permeability [31-33], although pathological reports have been infrequent and discordant [34]. Mechanistic insight provided by advanced neuroimaging methods suggests that there are likely early alterations in interstitial fluid mobility that eventually lead to demyelination and axonal damage [34]. Because WMH are highly prevalent in “healthy” populations (Table 1) [35-44], it remains unclear whether WMH are always pathologic. However, punctate WMH are probably due to a variety of causes and have relatively low risk for further progression. On the other hand, confluent WMH are likely to progress in a more aggressive fashion [45]. While not fully clear, subcortical WMH and periventricular WMH may represent the same spectrum of disease leading to devastating clinical outcomes [34,46]. Because of limitations in neuroimaging techniques and MRI processing methods, differentiating WMH from other lesions or artifacts (such as corticospinal tracts or blood flow) can be difficult [34].

Major population-based studies of WMH in healthy subjects
Several population-based studies have explored the risk factors associated with development of CSVD. The major studies include the Cardiovascular Health Study (CHS), Austrian Stroke Prevention Study (ASPS), Rotterdam Study (RS), and Atherosclerosis Risk in Communities Study (ARIC). While these studies have yielded several conflicting results, there are some patterns that have emerged. In the CHS [35,36,47-53], a longitudinal study of elderly (≥65 years) community-dwelling adults, approximately three-fourths of the study population exhibited periventricular rather than subcortical WMH and about one-fifth of population had equal distributions in both regions. Higher WMH burden was associated with older age, asymptomatic stroke on MRI, higher systolic blood pressure (BP), smoking, and female gender, with age showing the strongest correlation [35]. By contrast, the majority of WMH in the RS, a population based-cohort study that recruited nondemented individuals aged 60 to 90 years [37,54-57], were found in subcortical areas and to greater extent in frontal and parietal regions [37,38]. As shown in a substudy of the RS, arterial stiffness (as measured by the aortic pulse wave velocity) was associated with larger WMH volumes independent of cardiovascular risk factors [58]. Similar to CHS, WMH were also associated with increasing age. Regardless of age or anatomic location, women had a higher burden of white matter lesions (both in periventricular and subcortical areas) consistent with findings from the PROspective Study of Pravastatin in the Elderly at Risk (PROSPER) study [59,60] as well as CHS. This may be due to decreased estrogen after menopause allowing the brain to be more prone to hypoxia [37]. Interestingly, in a longitudinal substudy of PROSPER [59], deep WMH progression among women was twice that of men, whereas the increase in periventricular WMH was similar [60]. In the ARIC study, a prospective study that recruited middle-aged men and women [61], hypertension was associated with WMH [39,62]. For unclear reasons, blacks had a lower prevalence of WMH, but a higher prevalence of severe WMH [63]. In a follow-up study, blacks, who had a higher incidence of baseline hypertension, experienced more WMH progression than Caucasians [64]. Unlike many of the other population-based studies (in which blacks were less represented), this finding suggests that hypertension may influence CSVD progression. However, it is uncertain whether black race is independently associated with WMH burden or whether hypertension contributes indirectly. In addition to blacks, Asians have a high burden of CSVD [65,66], although the location of their WMH is similar to that of Europeans, but more strongly associated with age [67]. Similar to blacks, hypertension is also strongly associated with WMH burden in Asian populations [68].
In early longitudinal studies, baseline CSVD (lacunes and WMH) seems to have the strongest association with WMH progression, although there has been conflicting information regarding whether WMH progression occurs in periventricular or subcortical (deep) areas [69-73]. In follow-up studies examining the progression of WMH in the CHS, the risk of clinical stroke was increased among patients with high baseline WMH burdens and asymptomatic strokes [36,52,74]. Interestingly, with the exception of cigarette smoking and presence of an infarct on baseline MRI, the relationship between risk factors and WMH progression was dependent on low versus high WMH burden at initial scan (Table 1) [36]. Baseline WMH burden was also found to be important in the ASPS, a single-center population-based study that enrolled around 2,000 individuals in Graz, Austria, ages 50 to 75 [40,75]. In a 3-year period, subcortical WMH progression occurred in deep and subcortical areas, and was associated with diastolic BP as well as baseline WMH burden [40]. However, in a 6-year follow-up study, only baseline WMH grade was ultimately associated with WMH progression [41,76]. Similar to the ASPS, the RS showed that WMH progression occurs in subcortical areas compared to periventricular areas [38], a finding which was also replicated in the Leukoaraiosis And DISability study (LADIS) [72]. Interestingly, one Japanese study of neurologically healthy adults demonstrated that subcortical WMH were more associated with increased future risk of stroke than periventricular WMH, although periventricular WMH were associated with an increased risk of death [46]. In the RS, WMH progression was associated with a broad range of previously-identified risk factors [38,77] and in young patients and in patients without severe WMH at baseline, hypertension was strongly associated with lesion progression. Interestingly, a small percentage of subjects demonstrated WMH regression in subcortical areas (and to a much lesser extent periventricular areas), highlighting the dynamic nature of CSVD [27].
In the Framingham Offspring study, WMH were associated with an increased risk of symptomatic stroke, dementia, amnestic cognitive impairment, and death [78-82]. Similar studies have shown that WMH can modulate the progression from normal to mild cognitive impairment [83], with periventricular WMH playing a more critical role than deep/subcortical WMH [84,85]. In the RS, apolipoprotein (APOE) ε4 carriers, which have increased rates of Alzheimer’s disease [86], had higher WMH burdens, especially if carriers had comorbid hypertension. This has been corroborated by other studies relating APOE to WMH burden [87-89]. In the RS, 4.2% of subjects developed dementia, which was associated with a higher burden of periventricular WMH compared to subcortical WMH [90].
Recent small subcortical infarcts and lacunes
Because of advancements in neuroimaging that have led to improved detection of asymptomatic infarcts, the STRIVE consortium separated lacunar stroke into recent small subcortical infarct and lacune of presumed vascular origin [20]. Lacunes likely represent the chronic end-product of small subcortical infarcts. Recent small subcortical infarcts are sometimes accompanied by clinical lacunar syndromes, as initially described by Fisher [91,92], or they can be found incidentally by lesions with restricted diffusion on diffusion-weighted imaging (DWI) without an accompanying clinical stroke syndrome [93,94]. Small subcortical infarcts are dynamic, and over-time, can disappear, cavitate (forming lacunes), or form WMH [27,95,96]. On imaging, small subcortical infarcts follow the territory of a perforating arteriole, are <20 mm in maximal diameter, and exclude striatocapsular infarcts and anterior choroidal infarcts (which have a different underlying etiology). They are relatively easy to detect on MRI because they appear hyperintense on DWI and hypointense on apparent diffusion coefficient images. Pathologically, they are presumably due to occlusion of small perforating arterioles through a variety of mechanisms including microatheromatous disease or cardioembolism.
Lacunes of presumed vascular origin are appropriately named given the fluid-filled cavity that is often found after a lacunar stroke. By consensus definitions, lacunes are a round or ovoid, subcortical, fluid-filled cavity between 3 and 15 mm in diameter. They follow the signal intensity of cerebrospinal fluid on all sequences and on FLAIR images, and they have a central hypointensity with a surrounding rim of hyperintensity (in contrast to PVS) [20]. Incidental small subcortical infarcts are not commonly observed at high-frequencies in studies of asymptomatic populations, and so therefore our analysis will be limited to lacunes. Given that previous analysis of lacunes have been reviewed systematically [10,97], we will highlight only the major population-based studies.
In healthy populations, the frequency of lacunes ranges from 8% to 31% (owing to differences in the ages of the study populations and variability in spatial resolution of imaging studies) [21,41,51,52,56,68,98]. Although the associated risk factors have varied between studies, hypertension, age, and (to a lesser degree) smoking, seem to be the most replicated [21,56,68,98,99]. Conflicting associations between gender and infarct presence have been observed: in the CHS and Shunyi Study, male gender was found to be associated [21,68], and in the RS, female gender was found to be associated [56]. Other less robust risk factors that have been shown to be associated with silent lacunes include serum creatinine [21], diabetes [21,68,99], cholesterol [99], homocysteine levels [99], and black race [98]. Interestingly, carotid atherosclerosis was found to be associated with silent lacunes, suggesting that small subcortical infarcts can occur through artery-artery embolism or flow limiting effects [21,98,99].
In these studies, lacunes were typically found in the lentiform nucleus and thalamus [21] whereas a large portion of symptomatic infarcts were found in the cerebral cortex, thereby invoking other etiologies such as internal carotid artery atherosclerosis or atrial fibrillation [52]. Although many of these individuals did not experience any clinical symptoms of stroke, they were more likely to perform poorly on neuropsychological testing, highlighting the “covert” nature of CSVD [53]. In longitudinal studies, the progression of lacunes occurs in 3.5% [53] to 4.1% [99] of subjects annually. Baseline cerebrovascular disease (WMH and lacunes) seems to be the most prominent risk factor for development of additional lacunar infarcts, which are predominantly located in subcortical regions [53] or in the deep basal ganglia [56]. The association between baseline WMH and infarct development suggests that there may be a common pathway underlying these two markers. Similarly, symptomatic strokes are likely to represent a lesion in the same spectrum as asymptomatic strokes. Individuals with these silent brain infarcts are twice as likely to develop symptomatic stroke, which share many of the same risk factors as silent infarcts [52]. Of the individuals that experienced a symptomatic stroke during followup studies, the majority were more likely to have a silent lacunar stroke or a higher WMH burden on their initial MRI [100].
Cerebral microbleeds
CMBs are areas (≤10 mm) of round/oval shaped signal voids with blooming effect on MRI, best seen on T2*-weighted gradient-recalled echo (GRE) or susceptibility-weighted imaging [101]. They are located in the cortico-subcortical junction and deep gray or white matter throughout the brain (including brainstem and cerebellum) [20,102]. Histopathologically, in microangiopathies that affect small penetrating vessels (such as non-CAA CSVD/”hypertensive vasculopathy” or CAA), there is often extravasation of blood products into perivascular tissues leading to activation of macrophages; these foci of hemosiderin-laden macrophages are representative of microbleeds [103]. Clinically, they are associated with ICH and stroke (especially in CAA) [104]. However, the clinical significance of CMB in healthy populations is poorly understood.
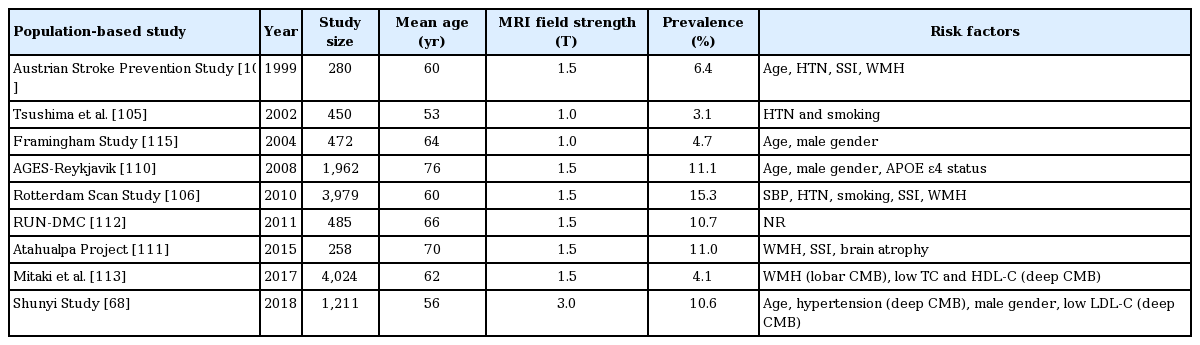
CMB are a common finding in “healthy” populations. The prevalence of CMB ranges from 3.1% [105] to 15.3% [106] owing to differences in the sensitivity of MRI sequences used (Table 2). In subjects with CMB, the majority have <3 lesions located in cortico-subcortical areas [107-112]. However, in non-European populations, CMB are found more predominantly in deep areas, which may be related to increased rates of hypertension although the prevalence of CMB is relatively the same [68,111,113,114]. While several risk factors are associated with CMB presence, the two most consistently associated are age and hypertension [68,105-107,110,111,115]. For example, in the Framingham Study Original Cohort and Offspring Cohort [108,109], the prevalence of CMB was 12.6% in patients 75 years of age or older compared to 2.2% in patients younger than 75. With improved detection sensitivity using 3DGRE in the RS, CMB prevalence was 18% in individuals aged 60 to 69 years and 38% in patients older than 80 [116]. In addition, three studies [68,110,115] have found higher rates of CMB in males, although other studies have not reported any gender differences in CMB prevalence [107]. As seen in other CSVD biomarkers, smoking was also identified as a risk factor in two studies [105,106] while diabetes and hypercholesterolemia may be less important (Table 2) [68,113]. Given the overlap of risk factors in CMB, WMH, and lacunes, there are likely shared underlying mechanisms that give rise to these biomarkers. Indeed, several studies have demonstrated an increased prevalence of CMB if concomitant WMH and lacunes were present [106,107,111,113].
Prevalence and risk factors of CMBs in healthy populations
As with other biomarkers, the location of CMB may be reflective of distinct underlying etiologies. In the ASPS, all individuals with deep CMB had hypertension, whereas only 50% of individuals with cortico-subcortical CMB had hypertension [107]. In the Framingham Study [108,109], almost three-fourths of the study population had CMB in the cortex, whereas 23% were located in deep regions. Unlike the aforementioned studies, hypertension was not associated with CMB, perhaps explained by the fact that this study included fewer subjects with deep CMB (of which hypertension is more related). In the Age Gene/Environment Susceptibility (AGES)-Reykjavik study [117], larger microbleeds were associated with hypertension, and unlike the ASPS, CMB presence was associated with ApoE ε4 homozygosity [110]. Similar to the Framingham Study, 70% of subjects had CMB located in cortex, whereas 11% had deep CMBs, and 19% had infratentorial CMB. Based on these findings, CAA seemed prevalent in their population, and given the association with ApoE, CAA was likely the etiologic culprit. From the RS [116], ApoE ε4 carriers had more lobar CMB compared to noncarriers, whereas patients with cardiovascular risk factors (hypertension, smoking), silent lacunar infarcts, and WMH had more deep or infratentorial CMB. These findings correlate with observed findings in ICH [118], and arterial stiffness findings in stroke patients [119]. Interestingly, in a follow-up analysis of the RS [120], it was shown that CMB were more prevalent (odds ratio [OR], 1.71) in patients taking antithrombotic medications (platelet aggregation inhibitors), but not in patients on anticoagulant medications. However, it is worth noting that CMB prevalence may be confounded by some indications for antiplatelet treatment, such as stroke [121]. Patients with strictly lobar CMB were found in a higher proportion among aspirin users (OR, 2.70) compared to carbasalate calcium users (a combination formula of calcium acetylsalicylate and urea; OR, 1.16), suggesting that aspirin worsens CMB burden in patients with CAA.
Even in clinically healthy persons, the presence of CMB correlates with numerous future risks. In the RS, the presence of CMB almost doubled the risk of ischemic stroke in a 5-year period, with greater CMB count associated with higher stroke risk [118]. In the PROSPER trial, subjects with greater than 1 CMB had a six-fold increase in stroke-related death than those without CMB [122]. Nonlobar CMB were associated with an increased risk of cardiovascular death whereas lobar CMB was associated with an elevated risk of stroke-related death [123]. CMB have also been associated with gait disturbances in nondemented persons [112,124]. Furthermore, subjects with ≥2 CMB performed poorly on tests of processing speed and executive function, especially those with multiple, deep or infratentorial CMB [125]. The presence of these deep CMB doubled the risk of vascular dementia (VaD). Subjects with both CMB and concomitant retinopathy were most likely to exhibit slow processing speeds, poor executive function, and VaD [125]. Other studies have shown that subjects with numerous (≥5) lobar CMB had more robust associations with cognitive dysfunction than did subjects with deep CMB [106,126]. However, in the Framingham Heart Study, strictly lobar CMB were not associated with an increased risk of dementia [127]. Collectively, these studies suggest that deep CMB may contribute to cognitive decline through mechanisms distinct from lobar CMB-associated cognitive dysfunction [126]. The former like reflects sporadic, non-amyloid CSVD whereas the latter reflects CAA.
Enlarged perivascular spaces
PVSs, sometimes referred to as Virchow-Robins spaces, are linear (but can be ovoid or round) projections that follow the path of blood vessels. Their significance is unclear, although they may be involved in the drainage of interstitial fluid and may be implicated in neurological diseases [128]. Normally, they are not well visualized by conventional MRI; however, as these spaces dilate (with age or other pathological conditions), they may be seen as fluid-filled spaces in both the gray and white matter [20]. They are usually <3 mm in maximal diameter, a cutoff that has often been used to distinguish these from lacunes, which also contain a hyperintense T2 rim surrounding the cavitation.
Similar to other biomarkers, the spatial distribution of PVS may reflect distinct mechanisms such that deep PVS are likely caused by hypertensive-CSVD whereas white matter PVS may be more driven by CAA. Such anatomical distinctions have also been observed in PVS found after ICH [129]. In patients presenting to a memory clinic, white matter PVS were associated with lobar CMB whereas basal ganglia PVS were associated with older age, hypertension, and higher WMH volumes [130]. Similarly, in a healthy Japanese cohort, basal ganglia PVS were associated with infratentorial CMB, and centrum semiovale PVS were associated with strictly lobar CMB [131]. Furthermore, basal ganglia PVS were associated with hypertension, lacunes, and severe WMH suggesting that these CSVD markers share common substrates. In the Three-City (3C)-Dijon Magnetic Resonance Imaging Study, in addition to the basal ganglia and white matter, PVS were also observed in the hippocampus and hypothalamus [132]. In this study, PVS were found in all subjects, with approximately one-third being large (>3 mm). Men appeared to have more basal ganglia PVS than woman, although this has not been replicated in other studies [133]. The severity of both basal ganglia and white matter PVS correlated with increasing age [134]. With increasing WMH volumes and lacunes, the odds of having more severe PVS in the basal ganglia was higher than in white matter.
While several of these studies employ 3 mm as a size cutoff to distinguish PVS from lacunes, in imaging-pathological correlation studies, an absolute cutoff size has not been established [135]. PVS above this size cutoff (termed large PVS) may actually be common and were found in approximately one-sixth of the subjects in the AGES-Reykjavik Study [117,136]. The majority of these large PVS were located in the basal ganglia and only a small fraction of subjects had exclusively white matter PVS. After controlling for age and sex, the presence of these deep large PVS was associated with silent subcortical infarcts and WMH progression. CMB were associated with both deep and white matter large PVS. The concurrent presence of PVS and other markers of CSVD suggests that there are common underlying mechanisms at play.
Although their significance in either healthy or diseased populations is not well understood, the presence of PVS may confer an increased risk of cognitive decline. In one small study, higher numbers of basal ganglia and centrum semiovale PVS were associated with poor performance on neuropsychological testing [137]. Independent of other cerebrovascular risk factors, large PVS (>3 mm) were associated with poor processing speeds as well as a four-fold increase in VaD risk, but not Alzheimer’s dementia (AD) or all-cause dementia risk [136]. This is consistent with previous studies that observed PVS in higher frequencies in patients with VaD compared to AD [138,139]. Furthermore, in another population-based study of elderly nondemented subjects [140], patients with the highest degree of both white matter and basal ganglia PVS were most likely to develop dementia over a 4-year period.
Cortical superficial siderosis
cSS are chronic hemorrhagic products that lie under the pia mater or in the subarachnoid space and are due to a variety of causes including small vascular malformations or CAA [20]. They are best visualized on paramagnetic MRI sequences given their hemosiderin composition, and appear as a linear hypointensity over the cortex.
In the RS, cSS were found in 0.7% of individuals, all of which had lobar CMB [141]. In these individuals, cSS was located near regions of lobar CMB (frontal and occipital areas). In the most recent Rotterdam data, the prevalence of cSS was 0.4% (n=3,401) [142]. These 15 individuals had concurrent CMB, the majority (80%) of which were located in lobar regions. In the 1,425 individuals who received a second MRI at 3 years, two subjects developed new cSS, one of which also developed a new lobar CMB. Of the seven that had baseline cSS, four showed progression of cSS on a follow-up scan. These individuals had several lobar CMB on baseline scans ranging from 7 to 130 CMB.
In another population-based study (n=1,412) [143] of individuals aged 50 to 89 (of which a portion had dementia or mild cognitive impairment [144]), using data from the Mayo Clinic Study of Aging, 13 subjects (0.9%) were found to have cSS. Unlike the RS, only about one-fifth of these patients had concurrent CMB, a finding which is consistent with other reports [145]. While the presence of the ApoE ε4 allele did not influence cSS burden, subjects with cSS were more likely to have the presence of the APOE ε2 allele, which is associated with CAA [146]. In a subset of patients who underwent Pittsburgh compound B (PiB) positron emission tomographic scans, those with cSS were more likely to be PiB positive (suggestive of high β-amyloid burden). Two patients with disseminated cSS had follow-up scans which demonstrated progression of both cSS and CMB. Although small numbers of subjects had both PiB and APOE data, these results suggest that cSS is intimately associated with CAA. Collectively, these findings suggest that cSS may be a marker of CAA-related CSVD rather than hypertensive arteriopathy. cSS is likely a rare finding in healthy populations but is probably exclusively related to coincident CAA pathology when it is found.
Cerebral microinfarcts
CMI are microscopic, presumed ischemic infarcts, mostly identified on pathological analysis, but can be seen on 7T MRI studies, and occasionally on 3T studies [147-150]. Given their ability to escape detection using standard neuroimaging protocols (1.5T MRI), CMI have not been evaluated extensively in healthy individuals. Because their small size (average of 0.2 to 1 mm in diameter) is below the spatial resolution (1 mm3) for most conventional MRI field strengths, higher strength MRI machines are needed to visualized them [147,151-153]. While most of the available data on CMI has come through post-mortem analysis by 7T MRI, recently, several groups have reported the ability to detect these lesions on 3T MRI in vivo [149,150,154-156]. However, the sensitivity of detection of 3T is limited compared to 7T [149], and can only detect “large” CMI (1 to 3 mm; whereas 7T can detect CMI <1 mm) [152]. As in vivo detection of CMI is relatively recent, it has not been incorporated into STRIVE definitions, and there have been no consensus definitions established. In some studies [149,150], CMI have been defined on 3T MRI as hypointense on T1-weighted images, hyperintense or isointense on FLAIR and T2-weighted images, distinct from PVS, <5 mm in maximum diameter, and perpendicular to the cortical surface.
In patients presenting to a memory clinic in Singapore, the prevalence of CMI determined by 3T MRI was 32% [149]. The presence of CMI was associated with hyperlipidemia, history of stroke, and cardiovascular disease (even after controlling for macroinfarcts). Furthermore, the presence of cortical CMI was associated with executive dysfunction as well as lower scores on neurocognitive testing with relative sparing of areas involved in subcortical disease (attention and visuomotor speed). Not surprisingly, patients with cortical CMI were more likely to be diagnosed with VaD than those without CMI, although previous reports have indicated that the prevalence of individuals with CMI was relatively the same between AD patients and non-demented controls [153,157]. CMI were associated with both deep and lobar CMB, increased WMH burden, and small brain volumes. Even after adjusting for WMH, CMB, or macroinfarcts, CMI were independently associated with VCI [149]. Furthermore, CMI were associated with cortical and subcortical macroinfarcts (and were not always restricted to the hemisphere of the infarct). CMI were also associated with intracranial stenosis of the vessel supplying the affected territory. These findings have been reproduced in stroke patients, in which it was determined that internal carotid artery stenosis was an independent risk factor for the development of CMI, and CMI development was associated with higher stroke recurrence rates [158]. Overall, this study of memory clinic patients suggests that CMI are a common finding in elderly patients with cognitive dysfunction and is in accordance with other population-based neuropathological studies [159]. This study also reported hyperlipidemia as a risk factor for CMI whereas previous population-based autopsy studies identified hypertension as the causative agent [160,161]. Interestingly, in another analysis using this Singapore cohort [162], 29% of subjects exhibited CMI on 3T MRI. These patients had an increased frequency of hypertension, hyperlipidemia, cardiac disease, and were found to have higher levels of subclinical cardiac biomarkers including N-terminal pro-brain natriuretic peptide (nT-proBNP) and high-sensitivity cardiac troponin T (hs-cTnT) (after adjusting for age, sex, cardiac disease, and other cerebrovascular risk factors). In addition, there was an association between atrial fibrillation, ischemic heart disease, and congestive heart failure with cortical CMI. Indeed, in one neuropathological study, it was suggested that CMI may be caused by microemboli [163]. The constellations of these associations including hyperlipidemia, intracranial atherosclerosis, and cardiac disease suggests that CMI have a heterogenous etiology that arises from shared pathways of CSVD, cardioembolism, and large vessel stenosis.
In another large, population-based study that recruited participants from the multiethnic Epidemiology of Dementia in Singapore Study (EDIS; n=1,598), 6.3% exhibited at least one CMI [164,165]. The majority of these were located in the parietal lobes (42%), and to a lesser extent, frontal (21%), occipital (12%), and temporal lobes consistent with one earlier small study using 7T MRI [157]. The presence of other markers of CSVD (including lacunar infarcts, WMH, and CMB) as well as macroinfarcts and intracranial stenosis was greater in patients with CMI. Risk factors associated with CMI were increasing age, Malay ethnicity, hypertension, diabetes, and history of stroke. Although not consistent with previous reports [166,167], the association of CMI with diabetes suggests that CMI may also be caused by microatheromatous disease at penetrating capillaries. While it was not significant in this study, cigarette smoking has been associated with the presence of CMI in other studies [168]. In addition to cerebrovascular risk factors, subjects with CMI were more likely to have moderate cognitive impairment and dementia [165]. Furthermore, the presence of CMI was associated with poor performance on neuropsychological testing even after controlling for lacunar infarcts. Collectively, CMI are independently associated with neurocognitive impairment as has been observed in neuropathological studies [169].
Very few studies have examined whether the anatomical location of CMI suggests different risk factors or underlying pathologies. In one recent large population-based neuropathological study of elderly community dwelling individuals (n=1,066) [170-172], CMI were found in one-third of subjects. The odds of having one or more CMI were increased in patients with atherosclerosis, arteriolosclerosis, and CAA. However, in a subgroup analysis, only atherosclerosis and arteriolosclerosis were associated with subcortical CMI, whereas CAA was associated with cortical CMI. Similar to other biomarkers, cortical CMI may be associated with CAA pathology whereas subcortical CMI may be due to other mechanisms such as hypertension.
Screening for CSVD
The major clinical features and associated risk factors for CSVD biomarkers are summarized below (Table 3). Currently, screening of asymptomatic individuals for CSVD is not recommended [173]. The major reasons MRIs are not performed in healthy populations is cost and lack of intervenable measures if asymptomatic CSVD is identified. However, more cost-effective methods of screening for CSVD may be able to select at-risk patients, especially those in high-risk populations, such as women or African Americans. These initial studies, if abnormal, can prompt further testing including MRI.

Summary of biomarkers
In patients that have received a brain MRI, a total SVD score can be calculated to assess the total burden of CSVD [174]. This visually rated score (ranging from 0 to 4) incorporates WMH, lacunes, CMB, and PVS, and reflects the amount of brain injury attributable to CSVD [175]. Not surprisingly, hypertension, age, male gender, and smoking are associated with higher SVD scores, highlighting the concept of shared underlying pathogenic mechanisms. Although it has not been widely adopted in clinical practice, this score can assist in stratifying patients at risk for ischemic stroke [176] or cognitive impairment. Furthermore, it can serve as a surrogate combined marker for CSVD in clinical trials focused on secondary prevention [174].
In cross-sectional population-based studies, numerous biomarkers such as fibrinogen, C-reactive protein (CRP), interleukin-6, neurofilament light chain, homocysteine, and D-dimer have been associated with the presence of CSVD markers such as WMH, lacunes, and PVS [177-179]. In two longitudinal populationbased studies, only intercellular adhesion molecule 1 (ICAM-1) and CRP have been found to be associated with WMH progression [180,181]. However, given that there have been conflicting results in these studies [182-185], the use of biomarkers for routine screening of CSVD is not recommended in healthy populations. These markers may be incidental, and their significance and progression remain unclear. Several of these biomarkers, such as homocysteine are not entirely specific for CSVD and can be elevated in other stroke subtypes [186]. Moreover, interpretation of cut off values and heterogeneity among studies confounds interpretation of results. Future efforts should be aimed at identifying specific markers for asymptomatic CSVD, markers that represent accelerated CSVD progression, and markers that predict conversion from asymptomatic to symptomatic CSVD.
Treatment and future directions
It remains unclear how to best identify patients with asymptomatic CSVD. Furthermore, it is unclear whether treatment of CSVD at an asymptomatic disease stage is necessary or beneficial [173]. Perhaps the most available data comes from studies on WMH in community-dwelling individuals. As hypertension seems to play the biggest role in CSVD pathogenesis, it is not surprising that the majority of these studies focus on hypertension reduction. Early trials have demonstrated higher WMH volumes in patients with uncontrolled hypertension compared to those receiving antihypertensive medications [187,188]. Furthermore, several trials have demonstrated reduced WMH progression with BP reduction therapy [189-193], although these results were not replicated in all studies [194]. When these trials were analyzed in a recent meta-analysis [193], less WMH progression was observed in subjects taking antihypertensive medications. The optimal timing for usage of these antihypertensives remains unclear, but presumably, earlier intervention may be more beneficial. Whether antihypertensive medications have similar effects on other CSVD biomarkers in unknown.
In addition to antihypertensives, other studies have examined statin usage, although very few population-based studies demonstrate an association of hyperlipidemia with CSVD. Three studies have demonstrated reductions in WMH with lipid-lowering drugs [195-197], while other studies did not show any benefit [198,199]. Although the association between diabetes and CSVD is controversial [200,201], hyperglycemia was shown to influence WMH progression in one study [72]. No studies, however, have evaluated glycemic control on WMH reduction. Lastly, in one meta-analysis, half of the studies demonstrated WMH reduction with exercise, but the other half did not [202].
Although antiplatelet agents have been extensively evaluated in the primary and secondary prevention of patients with lacunar stroke, there have been few studies examining the cerebrovascular effects of antiplatelet agents in healthy populations. In one small study, healthy subjects who underwent MRI screening for WMH were given antiplatelet therapy [203,204]. In a follow-up MRI study 5 years later, there was no difference in deep or periventricular WMH progression among patients taking antiplatelet agents. Given the small observational nature of this study, larger studies will need to be performed to fully elucidate the effects of antiplatelet therapy on CSVD prevention. However, it should be noted that these therapies may be counterbalanced by an increased risk of CMB (and subsequent ICH) [205,206].
Some patients with CSVD may exhibit minimal or no clinical symptoms while other patients develop stroke, cognitive impairment, and other long-term morbidity [6,7,27]. Early detection of clinically significant CSVD is particularly difficult for reasons including (1) utility of screening in the general population and (2) lack of full understanding of the significance of CSVD biomarkers [207]. Part of the challenge in developing a validated brain biomarker for CSVD is the complex pathways that underlie the disease. Risk prediction models to select populations at higher risk are urgently needed.
Notes
Disclosure
The authors have no financial conflicts of interest.
Acknowledgements
This work was supported by the National Institutes of Health (R01AG047975, R01AG026484, P50AG005134, and K23AG028 72605 to AV and R25NS065743 to RWR).