Cerebrovascular Drug-Eluting Stent versus Bare-Metal Stent in the Treatment of Vertebral Artery Stenosis: A Non-Inferiority Randomized Clinical Trial
Article information
Dear Sir:
Drug-eluting coronary stents have been used for patients with vertebral artery stenosis to prevent the occurrence of in-stent restenosis which affects the therapeutic efficacy of angioplasty and stenting. However, the results are inconsistent [1]. A new cerebrovascular sirolimus-eluting stent system (Maurora, Alain Biotechnology Co. Ltd., Beijing, China) [2], different from the Apollo stent systems (Apollo, MicroPort Scientific Corp., Shanghai, China) made of 316L stainless steel [3], adopts L605 cobalt-chromium alloy that offers a higher yield and has tensile strength characteristics allowing for thinner thickness while maintaining adequate strength and flexibility suitable for a curved artery of the brain, and the sirolimus can reduce the cell proliferation. This trial was to evaluate the safety and efficacy of this new drug-eluting stent.
This was a single center, open-label, prospective, non-inferiority, randomized, controlled trial (ChiCTR-IIR-16009115) [2]. Inclusion and exclusion criteria were based on the Endovascular Interventional Treatment for Ischemic Stroke Guideline of China (Table 1). Patients aged 18 years or older with intracranial vertebral artery stenosis of at least 70% and presence of transient ischemic attack or ischemic stroke were enrolled. And, for the patients with extracranial vertebral artery stenosis of at least 70%, presence of symptoms was not necessary. The sample size of this non-inferiority trial was calculated based on our previous retrospective data [4] and the non-inferiority margin [2] (△=6%). Patients were randomized in a 1:1 ratio to undergo stenting with Maurora stents or Apollo stents between September 2014 and September 2015.
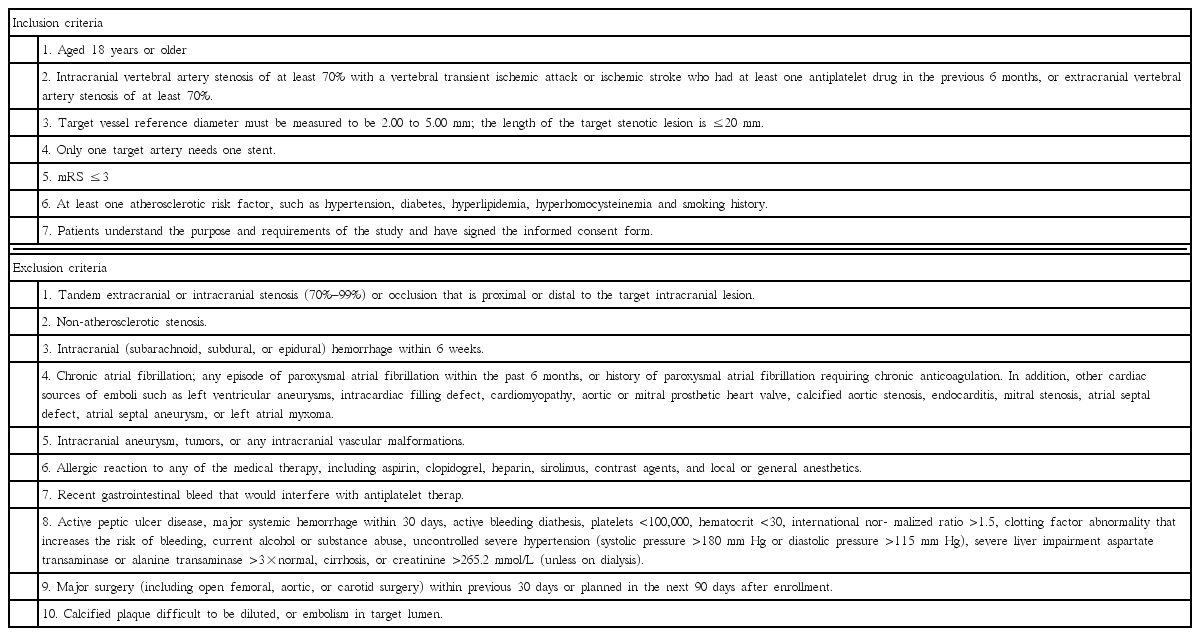
The inclusion and exclusion criteria
Primary outcomes included surgical complications within 30 days after procedure and the incidence of in-stent restenosis within 6 months after operation. Secondary outcomes included stroke ipsilateral to the target vertebral artery cerebrovascular and cardiovascular events, and serious adverse events within 12 months after operation. In-stent restenosis was defined as a lesion demonstrating more than 50% stenosis (within or immediately [within 5 mm] adjacent to the stent) and more than 30% absolute luminal loss at 6-month angiographic follow-up imaging (30% increase in posttreatment stenosis).
As a result, 40 enrolled patients were randomly divided into the two groups to receive stenting with either Maurora stent (drug-eluting stent group [DES-G], n=20) or Apollo stent (baremetal stent group [BMS-G], n=20), with no cross-over. Though the study groups were well balanced with regards to the baseline and procedure-related demographics data, which showed no significant differences (P>0.05), recent qualifying event (P=0.005) and location of target stenosis in the vertebral artery (P=0.008) showed statistical significance (Table 2). All the primary and secondary outcomes can be seen in Table 3. No procedure-related complications occurred within 30 days after procedure. The median angiography follow-up time was 6.5 months in DES-G and 6.4 months in BMS-G. In-stent restenosis rates were 5% in the DES-G and 25% in BMS-G with a difference of –20% (P=0.182), demonstrating non-inferiority (Figure 1).
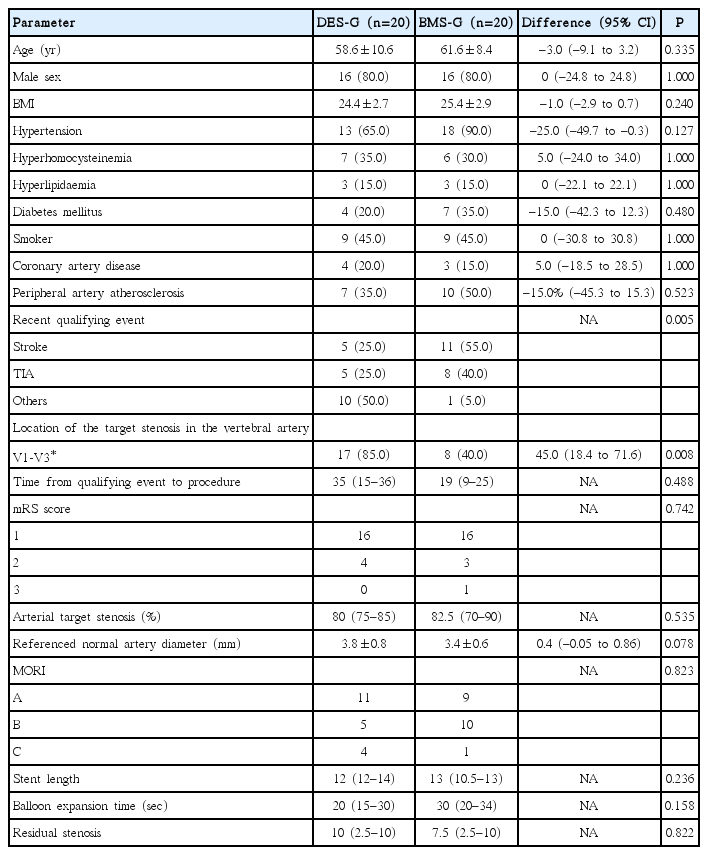
Baseline and procedure-related characteristics

Primary and secondary outcomes 30-day and 1-year after procedure
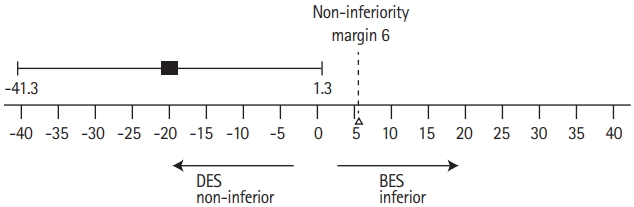
The non-inferiority text for restenosis. DES, drug-eluting stents; BMS, bare-metal stents.
The mean clinical follow-up was 18.0 months for DES-G and 18.8 months for BMS-G. Serious adverse events occurred in one patient in the DES-G and three in the BMS-G (P=0.605). The 1-year incidence rates for cerebrovascular and cardiovascular events were 5% in DES-G and 15% in BMS-G (log-rank test, P=0.317). The 1-year incidence rates for ipsilateral stroke were 0% in DES-G and 10% in BMS-G (log-rank test, P=0.152).
In this study, two strokes occurred all due to in-stent restenosis, which demonstrated that in-stent restenosis was one important factor that affected the stenting efficacy. Non-inferiority test used in the study reduced the the need of large sample size, but the basic characteristics between the two groups were not well balanced, and it failed to obtain a superior result in the decrease in the in-stent stenosis. However, as far as we know, this study was the first randomized controlled trial that used special cerebrovascular drug-eluting stent for treating vertebral artery stenosis. The results showed that the cerebrovascular drug-eluting stent for the treatment of vertebral artery stenosis was safe, and was not inferior to the bare metal stent in reducing the restenosis rate. Although statistically insignificant, it showed a tendency to reduce the incidence of restenosis (5% and 25%). This study has laid the foundation for phase III multicenter clinical trial in the future.