Emerging Roles of microRNAs in Ischemic Stroke: As Possible Therapeutic Agents
Article information
Abstract
Stroke is one of the leading causes of death and physical disability worldwide. The consequences of stroke injuries are profound and persistent, causing in considerable burden to both the individual patient and society. Current treatments for ischemic stroke injuries have proved inadequate, partly owing to an incomplete understanding of the cellular and molecular changes that occur following ischemic stroke. MicroRNAs (miRNA) are endogenously expressed RNA molecules that function to inhibit mRNA translation and have key roles in the pathophysiological processes contributing to ischemic stroke injuries. Potential therapeutic areas to compensate these pathogenic processes include promoting angiogenesis, neurogenesis and neuroprotection. Several miRNAs, and their target genes, are recognized to be involved in these recoveries and repair mechanisms. The capacity of miRNAs to simultaneously regulate several target genes underlies their unique importance in ischemic stroke therapeutics. In this Review, we focus on the role of miRNAs as potential diagnostic and prognostic biomarkers, as well as promising therapeutic agents in cerebral ischemic stroke.
Introduction
Stroke
Stroke is the major cause of adult physical disability and the second leading cause of death in the world [1,2]. Stroke is one of the most important and devastating of all neurological disorders, accounting for 5.5 million deaths annually, with 44 million physical disabilities worldwide [3]. The consequences of stroke injuries are profound and persistent, causing a high burden to both the individual patient and society because of their increasing incidence, the physical disability and mortality they cause, and their economic impact, mainly in low- and middle-income countries [4].
Ischemic stroke is responsible for 80% of all strokes, while hemorrhagic stroke accounts for 15% and the other 5% are due to unknown etiology [5]. In the present review we will discuss the pathogenic mechanisms related to ischemic stroke such as excitotoxicity, oxidative stress, inflammation and apoptosis, and how microRNAs may play a role in these pathogenic process. We will also investigate miRNAs that involved in the post-stroke recovery and repair pathways.
Pathophysiology of cerebral ischemia
Cerebral ischemia, which leads to brain dysfunction, results from cerebral artery occlusion that decreases cerebral blood flow, and its symptoms last for 24 hours or more [6]. During ischemic stroke, neurons are deprived of oxygen and energy, so that their normal metabolic substrates stop functioning in seconds and display signs of structural injury after only 2 minutes [7]. Immediately after ischemia, cellular energy-dependent processes fail and neurons are unable to sustain their normal transmembrane ionic gradient, resulting in an imbalance between ions and water thus leading to apoptosis and necrotic cell death [8,9].
During ischemia the brain tissues are not affected equally owing to differential lessening of blood supply to the different zones. Hence, ischemic injury involves the ischemic core and the penumbra region [10]. Severe ischemia occurs in the ischemic core, where neuronal damage is irreversible due to necrotic cell death while the surrounding penumbra constitutes cells that are metabolically active and potentially salvageable. Therefore, the penumbral zone has the potential for recovery and is the target for therapeutic agents [11,12]. Nevertheless, cerebral ischemia triggers several pathogenic processes (excitotoxicity, oxidative stress, inflammation and apoptosis) in the penumbra zone that leads to neuronal cell death (Figure 1). These processes are considered to be the central mechanisms underlying neuron death in ischemic stroke [13-15].
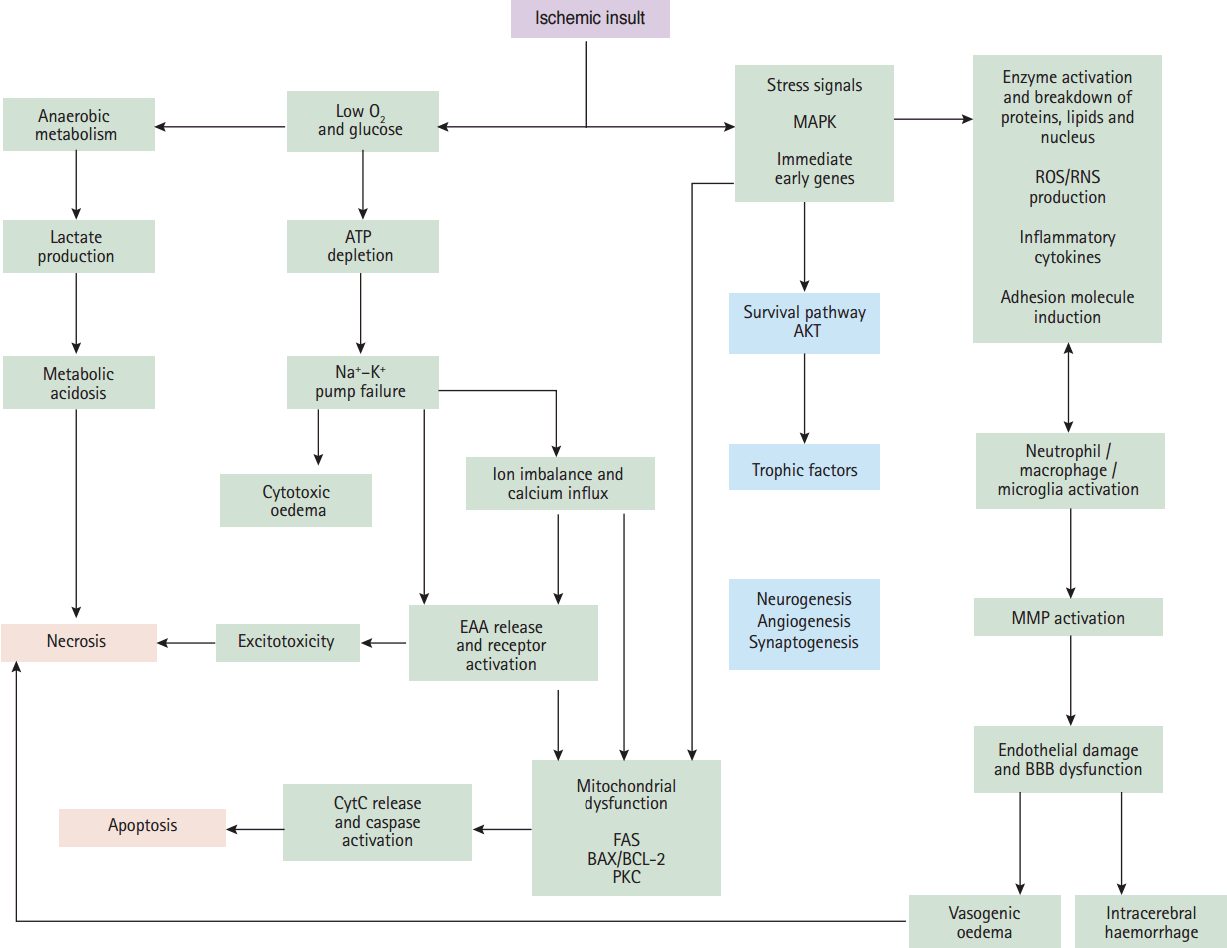
Critical events in the ischemic cascade. Following ischaemia, the deprivation of oxygen and glucose to the brain lead to loss of ATP (energy loss) and ion pump failure. The loss of ion concentration gradients causes cytotoxic oedema and releasing of excitatory amino acids (EAAs). Following reduced glucose availability cell aerobic metabolism switches to anaerobic, resulting in metabolic acidosis. All of these events lead to cell death, or necrosis. Ischaemia also causes the upregulation and activation of many immediate early genes and stress signals, which lead to inflammatory responses, cell apoptosis and, subsequently, activation of matrix metalloproteinases (MMPs) as a damaging protease which can lead to the brain oedema and haemorrhage. Following ischaemia, AKT kinase activation and upregulation of trophic factors set the stage for recovery and repair mechanisms which including neurogenesis, synaptogenesis and angiogenesis. AKT, protein kinase B; MAPK, mitogen-activated protein kinase; ROS/RNS, reactive oxygen species/reactive nitrogen species; ATP, adenosine triphosphate; EAA, excitatory amino acids; CytC, cytochrome c; FAS, the cell-surface Fas receptor; PKC, protein kinase C; BBB, blood brain barrier.
MicroRNAs
miRNAs are small, non-protein-coding RNAs, which include ~20–24 nucleotides that are highly conserved through evolution. They are post-transcriptional regulators that targeting the 3′-untranslated regions (3′-UTRs) of target mRNAs, which lead to the inhibition of translation or degradation of the respective mRNA [16]. miRNAs have been implicated in the regulation of a variety of cellular processes and diseases such as neuronal development, differentiation, synaptic plasticity, proliferation, metabolism, apoptosis, neurodegenerative diseases and tumorigenesis [17-22]. miRNAs are initially transcribed from genomic DNA, and RNA polymerase II is responsible for transcription of primary miRNA (pri-miRNA) [23]. Pri-miRNAs can be thousands of base pairs in length and consist of at least one hairpin loop, which is recognized and cleaved by the endonuclease Drosha, and which generates a precursor miRNA (pre-miRNA), with the help of DGCR8, a double stranded RNA-binding protein [24,25]. The pre-miRNA is transported from the nucleus into the cytoplasm through the function of exportin-5. In the cytoplasm, the premiRNA undergoes cleavage by endoribonucleic Dicer to form a duplex of the mature miRNA strand, which is generally biologically active [26-28].
It is known that biological functions of miRNAs are extremely dependent on the cellular context and the precise link between miRNAs and stroke consequences should be discussed only within a specific cellular context. The studies showed that miRNAs have participated as key mediators in the molecular processes underlying cerebral ischemia and related diseases [29-32]. Therefore, in the present study, we review all available relevant articles regarding miRNAs and ischemic stroke in order to explain the complex link between miRNA and ischemic stroke. The information about the stroke-miRNA system may be used for therapeutic and diagnostic methods in stroke treatment.
MicroRNAs intervention in ischemic stroke progression
In the past few decades, the clinical methods such as computed tomography scans and magnetic resonance imaging have facilitated diagnosis and prognosis of stroke. However, the diagnostic and prognostic powers are limited in availability and higher cost [33,34]. Additional diagnostic tools including interleukin-6 (IL-6), matrix metallopeptidase 9 (MMP-9) and C-reactive protein (CRP), which their specificity and ability to distinguish between acute stroke and its related risk factors is unclear [35]. Given the limited recommended therapeutic window for thrombolysis, new biomarkers are necessary for advancing diagnosis of stroke. Therefore, recent studies have suggested promising mRNA based biomarkers, which they could distinguish transient ischemic attack from control samples [36]. Hence, several studies have reported the uses of miRNAs as circulating biomarkers for diagnosis or prognosis of stroke (Table 1).

Overview of circulating miRNAs and their relationship with stroke
Several pathogenic processes are involved in ischemic stroke progression which include excitotoxicity, oxidative stress, inflammation and apoptosis [37]. The miRNAs discussed below regulate genes in these pathogenic processes by downregulating the gene expression (Figure 2, Table 2).
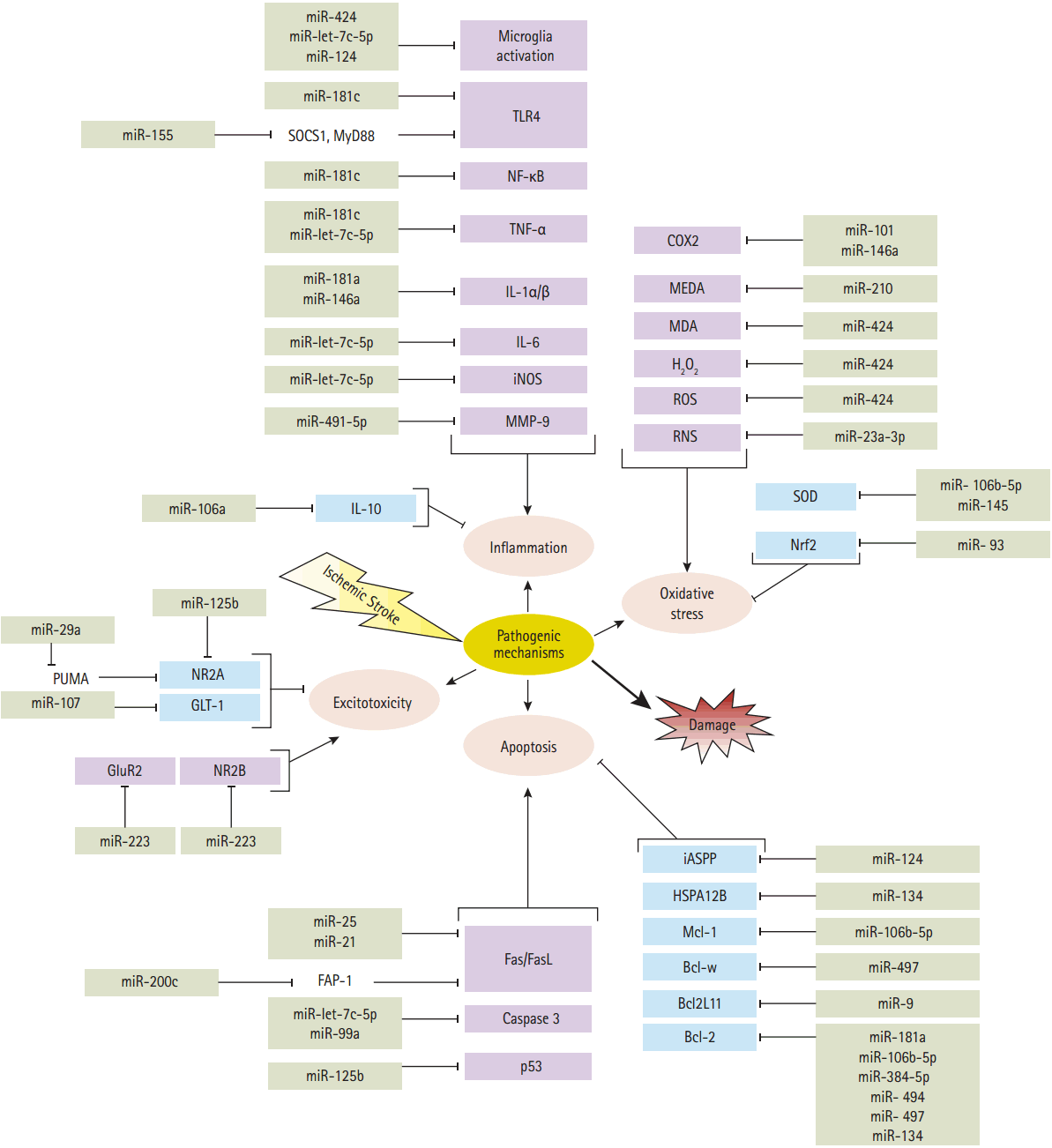
MicroRNAs involved in detrimental (purple boxes) and protective pathways (blue boxes) are activated by ischemic stroke. Cerebral ischemia, while activating detrimental pathways, also triggers some organized responses that counteract tissue injury. Post-ischemic oxidative stress triggers an oxidant and antioxidant responses via different factors which are inhibited by microRNAs. Oxidative agents that are inhibited by microRNAs, including reactive oxygen/nitrogen species (ROS/RNS), cyclooxygenase 2 (COX2), hydrogen peroxide (H2O2), malondialdehyde (MDA) and methane dicarboxylic aldehyde (MEDA). The antioxidant response which is inhibited by microRNAs containing transcription factor Nrf2 and superoxide dismutase (SOD). Following ischemia, inflammation is increased by production of matrix metalloproteinases (MMP-9) to infiltrate the BBB, and activation of pro-inflammatory genes such as interleukin-1 (IL-1α and IL-1β), IL-6, tumor necrosis factor α (TNF-α) and nuclear factor-κB, (NF-κB), as well as an activation of innate immune responses (microglia cells) and toll-like receptors (TLR4). Inflammation is mitigated by production of anti-inflammatory cytokines like such as IL-10. microRNAs could affect post-ischemic inflammatory and anti-inflammatory factors. Excitotoxicity associated with glutamate receptor activation can be counterbalance via glutamate transporter (GLT1) and NMDA (containing subunit NR2A), while glutamate receptors GluR2 and NMDA (containing subunit NR2B) exacerbate excitotoxic injuries. microRNAs inhibit those factors that contribute in the excitotoxicity. The detrimental effects of post-ischemic apoptosis are antagonized by activation and expression of antiapoptotic factors such as; Bcl-2, Bcl2L11, Bcl-w, Mcl-1 and the heat shock proteins family (HSPA12B). Hence, deleterious effects of apoptosis are induced by expression of caspase 3, activation of cell surface death receptors (Fas) and its ligand (FasL), and activation of p53, inhibitory member of the apoptosis-stimulating proteins of the p53 family (iASPP). There are some microRNAs which modulate the detrimental effects of post-ischemic apoptosis. SOCS1, suppressor of cytokine signaling 1; MyD88, myeloid differentiation primary response gene 88; iNOS, inducible nitric oxide synthase; Nrf2, nuclear factor erythroid-2 related factor 2; PUMA, p53 upregulated modulator of apoptosis; GLT-1, glutamate transporter-1; GluR2, glutamate receptor-2; FAP-1, Fas associated protein-tyrosine phosphatase 1.

Specific target genes of miRNAs involved in ischemic stroke pathogenesis
Post-ischemic excitotoxicity
Ischemic stroke damages brain tissue primarily through excitotoxicity, a term used to describe cell death induced by synaptic high levels of glutamate, which is a major excitatory neurotransmitter in the central nervous system (CNS) [38]. Several types of glutamate receptors have been identified in the CNS, and the three main types of these receptors are: α-Amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA) receptors, N-Methyl-D-aspartate (NMDA) receptors and metabotropic glutamate receptors (mGluR) [39-41]. Excess glutamate over-activates NMDA and AMPA receptors on postsynaptic cells which facilitate influx of calcium ions into neurons [37,42].
Under basal synaptic transmission, activation of the synaptic NMDA receptors (predominantly NR2A-containing) stimulates the signaling components of the neuronal survival signaling complex (NSC) that promoting neuronal survival [43,44]. However, under pathological conditions such as stroke, elevating of the extracellular glutamate concentration causing excitotoxic activation of extrasynaptic NMDA receptors (predominantly NR2B-containing). The NR2B activation increased Ca2+ influx and promotes active death-associated protein kinase (aDAPK) to bind with NR2B [43-45]. aDAPK recruitment promotes activating the neuronal death-signaling complex (NDC), that in turn suppress synaptic NSC activity [43], and mediate neuronal death. It is demonstrated that inhibition of aDAPK binding to the NR2B reduces activation of NDC and prevent the excitotoxic neuronal injury induced by ischemic stroke [45,46]. So, the NR2B subunit is a major hub for NDC formation [45,47,48].
Also, binding of glutamate to mGluR caused release of the intracellular calcium store [37,39]. These events result in accumulation of intracellular calcium which changes the osmolarity of the cell and activation some of endogenous enzymes such as proteases, lipases and endonucleases. These enzymes degrade important cellular macromolecules such as structural proteins, membrane lipids and DNA [37,39,49].
MicroRNAs and ischemic excitotoxicity
Following ischemic stroke, overexpression of miR-107 leads to suppression of glutamate transporter-1 (GLT-1) expression and elevated glutamate accumulation, which determine the degree of excitotoxicity [50]. Post-ischemic downregulation of GLT-1 is closely associated with accumulation of glutamate, suggesting that glutamate accumulation and neuronal excitotoxicity can be controlled via GLT-1 expression [51]. After transient forebrain ischemia, increasing miR-29a protects astrocytes and then indirectly neurons. miR-29a leads to decreasing PUMA (p53 upregulated modulator of apoptosis) levels and thereby preserves astrocyte GLT-1 leading to attenuation of oxidative stress and survival of neurons [52].
Overexpression of miR-223 attenuates NMDA-induced calcium influx in hippocampal neurons and protects the ischemic brain from excitotoxic neuronal cell death through suppression the levels of the glutamate receptor-2 (GluR2) and NMDA subunit NR2B [53]. It has been reported that the NR2A is a target for miR-125b and this miRNA negatively regulates NR2A expression [54]. It has been approved that activation of NR2B-containing NMDA receptors leading to excitotoxicity and apoptosis. While, activation of the NR2A-containing NMDA receptors exerts a neuroprotective effects and promotes neuronal survival against excitotoxic-mediated neuronal damage [44]. Synaptic plasticity that is profoundly influenced by the NMDA receptor subunit is altered [54]. This is a devastating effect because, after stroke damage plasticity can promote adult brain recovery [55-58].
Post-ischemic oxidative stress
Oxidative stress results from increased reactive oxygen/nitrogen species (ROS/RNS) and/or decrease of the anti-oxidative stress defense systems of the body [59]. Several mechanisms caused formation of free radicals and ROS during ischemia [60], including high stimulation of NMDA glutamate receptors due to excitotoxicity [61], Ca2+ overload, mitochondrial dysfunction [62-64], neuronal nitric oxide synthase (nNOS) activation [65], and migration of inflammatory cells such as neutrophils and leukocytes that can generate superoxide anions [66]. Oxidative stress has been involved in a variety of diseases, including cancer, atherosclerosis, neurodegenerative diseases, and stroke [67]. Oxidative damage is a fundamental mechanism of brain damage and neuronal cell death during ischemic stroke. The brain is very susceptible to oxidative stress due to its highly oxygenated environment, with high levels of peroxidisable lipids, low levels of antioxidants and a high iron content [68].
The activity of antioxidant and detoxifying enzymes such as superoxide dismutase (SOD), glutathione peroxidase, glutathione reductase and Glutathione-S-transferase (GST), has been studied in stroke patients, and these enzymes maintain redox homeostasis and influence the inflammatory response [69,70]. SOD enzymes (manganese SOD [MnSOD] and extracellular SOD) help brain recovery following ischemic reperfusion injuries [71,72]. The genes that encode these antioxidant enzymes bear an antioxidant response element (ARE) within their promoters. The transcriptional activation of ARE is mainly regulated by nuclear factor erythroid-2 related factor 2 (Nrf2) [73]. It has been determined that Nrf2 has a neuroprotective activity against stroke injuries, such as oxidative glutamate excitotoxicity, hydrogen peroxide (H2O2) exposure, and Ca2+ overload situations [74]. Moreover, Nrf2 expression is upregulated at the gene and protein levels in ischemic brains especially in the ischemic penumbra zone; these findings indicate that Nrf2 activation is valuable and might subsequently contribute to cell protection and survival [75].
MicroRNAs and ischemic oxidative stress
miRNAs have been observed to be involved in the posttranscriptional regulation of Nrf2 levels. It is discovered that 85 miRNAs can bind to cytoplasmic Nrf2 mRNA to affect its translation [76]. Studies demonstrated that miR-424 reduced malondialdehyde (MDA) levels, ROS and abrogated H2O2-induced injury in neurons which resulted in the neuroprotection against ischemic oxidative damages [77]. Evaluation the role of miR-93 in cerebral ischemia injuries indicate that miR-93 directly binds to the predicted 3′-UTR target sites of the Nrf2 genes, and then attenuate the expression of Nrf2 and heme oxygenase-1 (HO-1) [78]. The Nrf2/HO-1 pathway is an important cellular defense mechanism against oxidative stress induced following ischemia/reperfusion [79]. Also, it is revealed that increasing of Nrf2 levels causes upregulation of SOD enzymes [80,81].
Recent studies showed that vagus nerve stimulation (VNS) initiated after ischemic stroke in rats which improved the neurological outcomes, reduced ischemic lesion volume, and inhibited inflammatory cytokines [82]. It is known that miR-210 is involved in the VNS-regulated oxidative stress responses following cerebral ischemia through decreasing methane dicarboxylic aldehyde levels and increasing SOD and GSH levels [83]. In addition, ischemic stroke caused to the down-regulation of SOD and GSH activity and the up-regulation of methane dicarboxylic aldehyde [83]. It has been determined that acute ischemic stroke caused a significant increase of miR-106b-5p. Therefore, miR-106b-5p antisense oligonucleotides (antagomirs) could have a protective effect against post-ischemic oxidative damages via reducing MDA content and restoration of MnSOD activity [84]. miR-145 expression suppressed protein levels of SOD2 after ischemic stroke [31]. miR-23a-3p levels increased transiently following ischemia and reperfusion in mice which reduced the ischemia reperfusion and oxidative stress injuries, mechanistically through increasing the expression of MnSOD, and reducing RNS production such as NO and 3-NT levels [85].
During cerebral ischemia, cyclooxygenase 2 (COX2) can produce ROS [86]. COX2 is a qualified target of miR-101 [54]. In the normal situation COX2 is little expressed while studies showed that cerebral ischemia readily induced COX2 expression in neuronal cells [87]. The miR-101 profile in cerebral ischemia is found to be down-regulated [32]. Also, miR-146a has been found to suppress expression of COX-2 in neurological disorders [88]. Thus, miRNAs can be considered as a valuable therapeutic agents to antagonize oxidative stress in ischemic stroke.
Post-ischemic inflammation
Inflammation is an essential step and a secondary injury mechanism in the pathophysiology of cerebrovascular diseases, particularly ischemic stroke [89,90]. Recent studies demonstrate that post-ischemic neuro-inflammation is an important determining factor for ischemic consequences and its long-term prognoses [91,92]. In ischemic brain injury, inflammatory responses are triggered as a result of damaged tissue, necrotic cells, debris and ROS. These triggering elements cause microglial activation and release of inflammatory cytokines [93-96]. Microglia are the resident innate immune macrophages of the CNS, and they are highly activated after brain insult [97-99]. Activated microglia and their inflammatory factors, such as tumor necrosis factor α (TNF-α) contribute to the progression of neurodegenerative disorders [100,101].
Cytokine release leads to post-ischemic inflammation and aggravates primary brain damage. They include, IL-1β, IL-6, plasma high sensitivity CRP (hs-CRP) and TNF-α, as well as other potential cytotoxic molecules including NO, ROS, and prostanoids [102-105]. Microglial suppression can reduce post-ischemic injuries, so this illustrates an attractive therapeutic strategy for ischemic stroke [106,107]. In addition to cytokines that are expressed in the resident brain cells, there are a peripherally derived cytokines that produce and secrete from T-lymphocytes, mononuclear phagocytes, NK cells and polymorpho-nuclear leukocytes which are involved in ischemic inflammation [108].
In ischemic brain injury, the expression of a number of pro-inflammatory genes is induced by ROS formation. These genes include nuclear factor-κB (NF-κB), interferon regulator factor 1, hypoxia inducible factor 1 (HIF 1) and STAT3. Consequently, these factors upregulate cytokines and expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), P-selectin and E-selectin. These Cellular adhesion molecules (CAMs) facilitate leukocyte adhesion to the microvascular endothelium in the cerebral ischemic area [107]. NF-κB is a heteromeric transcription factor involved in the activation of pro-inflammatory genes, such as TNF-α, ICAM-1, COX-2, iNOS and IL-6 [109,110]. CAMs are upregulated in the first days of ischemic stroke and are responsible for the migration of the leukocytes through the brain endothelial cells [111].
During ischemia, neutrophils that are recruited to the ischemic tissue produced the MMPs to infiltrate the blood brain barrier (BBB). Two main group of MMPs are including MMP-9 and MMP-2, and they are responsible for disruption of BBB and hemorrhagic transformation following ischemic stroke [112,113].
MicroRNAs and ischemic inflammation
It has been clarified that a number of miRNAs target several genes that are involved in post-ischemic inflammation [114,115]. Studies showed that miR-424 has a protective effect against ischemic cerebral injuries by mechanisms that inhibit microglia activation [116]. Also, miR-let-7c-5p have a protective effect against cerebral ischemia neuro-inflammation via inhibition of microglial activation and translational repression of caspase 3 [117]. Overexpression of miR-124 could promote quiescence of microglia and deactivation of macrophages via the C/EBP-α-PU.1 pathway. miR-124 expression in the microglia was lessened during the neurological disease [118].
Ischemic inflammatory process may be the resulted of activation of Toll-like receptors (TLRs). TLRs are a family of receptors that are expressed by microglia and astrocytes [119-121]. TLRs can activate NF-κB which induces the expression of pro-inflammatory genes, cytokines and adhesion molecules [122]. Thirteen TLRs have been identified, and TLR4 signaling contributes to post-ischemic inflammatory injuries [123,124]. In response to hypoxia, TLR4 expression is upregulated in the surface of microglia cells [125]. It is determined that miR-181c negatively regulates TLR4 expression through its 3′-UTR. Furthermore, miR-181c suppresses NF-κB activation and its pro-inflammatory products including TNF-α, IL-1β, and iNOS [126]. In ischemic cerebral tissue, miR-155 induces the expression of TNF-α and IL-1β via upregulation of TLR4 and downregulates the expression of inflammatory mediators such as suppressor of cytokine signaling 1 (SOCS1) and the myeloid differentiation primary response gene 88 (MyD88) [127]. In the microglia, macrophages and monocytes, expression of the miR-155 was upregulated in response to the pro-inflammatory stimuli such as IFN-γ and TNF-α [128-130].
It has been shown that miR-181c can directly regulate post-transcriptional production of TNF-α in the microglia. Therefore, miR-181c decreased release of TNF-α from the microglial cells and decreased neuronal apoptosis [131]. Also, recent studies suggest that miR-let-7c decreases the expression of macrophages inflammatory genes including iNOS, TNF-α and IL-6 [132]. miR-181a has an anti-inflammatory effect via direct downregulation of IL1-α in monocytes and macrophage cell lines [133]. miR-146a has been found to suppressed expression of IL-1β and IL-6 which are pro-inflammatory cytokines. This finding indicates an important role of miR-146a in an inflammation associated with neurological disorders [88]. During cerebral ischemia, miR-146 is down-regulated [32]. anti-inflammatory cytokines such as IL-10 post transcriptionally regulated by miR-106a [134]. Moreover, in the microglia and macrophages miR-106a and miR-124 leading to increasing in IL-10 and TGF-β respectively [118,134,135]. Other findings indicate that serum miR-124, miR-9 and miR-219 were decreased in acute ischemic stroke thus the neuro inflammatory response and neuronal cell death was facilitated [136]. miR-491-5p was indicated to decrease the levels of MMP-9 expression and inhibit cellular invasion [137]. So, correlations between serum levels of miR-124, miR-9, miR-219, hs-CRP, MMPs and infarct volume in the acute phase of stroke were determined [136,138,139].
Post-ischemic cell death
Apoptosis, necrosis and necroptosis are three types of cell death involved in ischemic stroke pathogenesis. Apoptosis is programmed cell death and it is well known to be activated during development, physiological cellular turnover, and in pathological conditions such as stroke [140-142]. The apoptotic response is activated either by extrinsic or intrinsic stimuli; the intrinsic stimuli triggered through the mitochondrial signaling pathway; the extrinsic stimuli activated via cell surface death receptors, including TNF-α, Fas (CD95/APO1) and TNF related apoptosis inducing ligand (TRAIL) receptors [143,144]. The extrinsic pathway is activated by ligand-receptor interactions via the external signal. Ligands such as TNF-α and Fas ligand (FasL) bind to TNF-receptor and Fas receptor (FasR) respectively which initiates formation of death inducing signaling complex and caspase-3 activation [145]. Both pathways are interface at the point of caspase-3 activation which results to the mitochondrial membrane permeabilization, chromatin condensation, DNA fragmentation, and eventually cell death [146].
Cerebral ischemia caused cytotoxic accumulation of intracellular Ca2+ through the stimulation of NMDA and AMPA glutamate receptors. Increased intracellular calcium activates calpains resulting in the cleavage of Bcl-2 interacting domain to truncated Bid (tBid) [147]. At the mitochondrial membrane, tBid forms heterodimers by interaction with pro-apoptotic proteins such as Bad-Box and opens the mitochondrial transition pores which promote releasing of mitochondrial cytochrome c (Cytc) or apoptosis inducing factor (AIF) [148]. The released Cytc in the presence of adenosine triphosphate (ATP)/deoxy ATP binds to the apoptotic protease activating factor 1 and procaspase-9 to form an apoptosome which activates caspase-9 and subsequently caspase-3. Activated caspase-3 cleaves nDNA repair enzymes, which leads to nDNA damage and apoptotic cell death. Furthermore, AIF is translocated to the nucleus and initiates large-scale (50 kb) DNA fragmentation and cell death in a caspase-independent manner [149]. After focal ischemic stroke caspase activation is present in the penumbra zone, an ischemic high risk area, and hence inhibition of caspase can protect against focal ischemia injuries [150].
MicroRNAs and ischemic apoptosis
Several studies showed that expression and function of specific miRNAs could regulate post-ischemic neural death by altering the expression of the target genes [151,152]. miR-25 could modulate cerebral ischemia/reperfusion damage by downregulation of the Fas/FasL Pathway and apoptosis inhibition [153]. miR-29 was found to repress expression of Fas associated protein-tyrosine phosphatase 1 which is the inducer of the FasRs [154]. miR-29 was demonstrated to be up-regulated during cerebral ischemia in rat models [32]. Some evidence showed that miR-21 can target Fas-ligand and protect neurons from apoptosis during ischemia [155]. Fas/FasL belong to the TNF receptor/ligand superfamily of co-stimulatory molecules and play an essential role in the induction of apoptosis [156].
miR155 regulates various functions of cells and its knock-down could modulate apoptosis via regulating caspase-3 gene expression [157]. Other findings indicate that miR-99a suppressed both pro-caspase-3 and activated caspase-3 expression as well as preventing neural apoptosis following cerebral ischemic stroke [158]. miR-let-7c-5p has been reported to repress caspase 3 that led to protective effects against cerebral ischemia [117]. miR-9 expression could specifically regulate Bcl2L11 translation which led to decreasing cell apoptosis, also miR-9 is able to restore the neurological scores and behavioral abnormalities. However, in the ischemic brain, miR-9 expression was downregulated and reversing its level could rescue the abnormalities and cell apoptosis [159]. In response to different stimuli, Bcl2L11 is produced and can induce apoptosis by inactivating anti-apoptotic Bcl2 proteins and activating BAX-BAK1 [160,161]. Bcl-2 and Bcl-xl proteins are a key regulators in lessening post-ischemic apoptotic and cell death [162].
Following acute ischemic stroke, miR-106b-5p increased significantly and directly target the Mcl-1 protein which is a member of Bcl-2 family and a key regulator of apoptosis after DNA damage [84]. So, miR-497 increased ischemic neuronal cell death by negatively regulating anti-apoptotic proteins, such as Bcl-2 and Bcl-w [152]. Several reports demonstrated that miR-181a levels, decreased Bcl-2 proteins and increased evidence of astrocyte dysfunction [163]. Expression profiles of microRNAs following cerebral ischemia suggest that differentially expressed miRNA-384-5p and miRNA-494 caused Bcl-2 to significantly decreased [164]. Moreover, Downregulation of miR-134 alleviates ischemic injury through enhancing the Bcl-2 expression in neurons following oxygen glucose deprivation [165]. It is reported that miR-134 plays a critical role in the post-ischemic apoptosis and cell death through negatively modulating HSPA12B protein expression in a posttranscriptional manner [166]. HSPA12B is a member of the HSP70 family, and overexpression of this protein decreased apoptosis in the ischemic brain tissue [167,168]. Furthermore, it has been demonstrated that down-regulation of the miR-125b expression caused increasing p53 expression which acts as apoptosis mediator by the intrinsic pathway. In ischemic rats, miR-125b is down-regulated following reperfusion [169]. Also, miR-124 can downregulate the inhibitory member of the apoptosis-stimulating proteins of p53 family (iASPP), and promotes neuronal apoptosis after cerebral ischemia. Therefore, suppression of miR-124 could be a novel mechanism for non-transcriptionall regulation of neuronal apoptosis in focal cerebral ischemia [170]. ASPP family consists of 3 members: ASPP1, ASPP2, and iASPP, because they bind to the proteins such as Bcl-2 and RelA/p65 as key players in controlling apoptosis [171]. As mentioned in the previous paragraphs, VNS improved the neurological outcomes and reduced ischemic lesion volume after cerebral ischemia in rats. Therefore, VNS exerts neuroprotective effects against ischemic injuries potentially through anti-apoptotic activity of miR-210 which is mediated by hypoxia-inducible factor and Akt-dependent pathways [83]. Following brain ischemia the up-regulation of miR-323 promoted apoptosis and suppressed survival, whereas the inhibition of miR-323 could be a good agent for the prevention and therapy of cerebral ischemic injury [172]. Consequently, these microRNAs maybe involved in neuronal apoptosis during stroke.
MicroRNAs as possible therapeutic agents
The underlying pathophysiology of stroke is highly complicated, consisting of numerous pathological processes such as excitotoxicity, oxidative stress, inflammation and apoptosis. Currently, effective treatment for ischemic stroke is limited to recombinant tissue plasminogen activator (tPA) [173]. tPA is the only appropriate thrombolytic agent available for acute ischemic stroke treatment [174,175]. However, tPA is limited by its narrow therapeutic window, which can only be given up to 6 hours after onset of stroke, therefore, making it suitable to only a minority (less than 10%) of stroke patients [176]. Also, beside its beneficial thrombolytic role, tPA has deleterious effects including intracranial hemorrhage [177], and neurotoxicity [178]. In addition, studies showed that inhibition of tPA with plasminogen activator inhibitor-1 or neuroserpin have neuroprotective effects against ischemic brain damage [179,180]. Other alternative treatments include the use of other anti-thrombotic agents, mechanical thrombectomy and anti-platelet agents such as aspirin [181]. As noted, there are many limitations of thrombolytic treatment for stroke. Therefore, there is continuing research for novel therapeutic agents. miRNAs have remarkable potential as they are endogenous molecules that are capable of controlling the expression of potentially deleterious genes. Furthermore, miRNAs can regulate the genes that contribute in the neuroprotection, neurogenesis and angiogenesis which leading to enhancing recovery and repair mechanisms in ischemic stroke patients (Figure 3, Table 2).
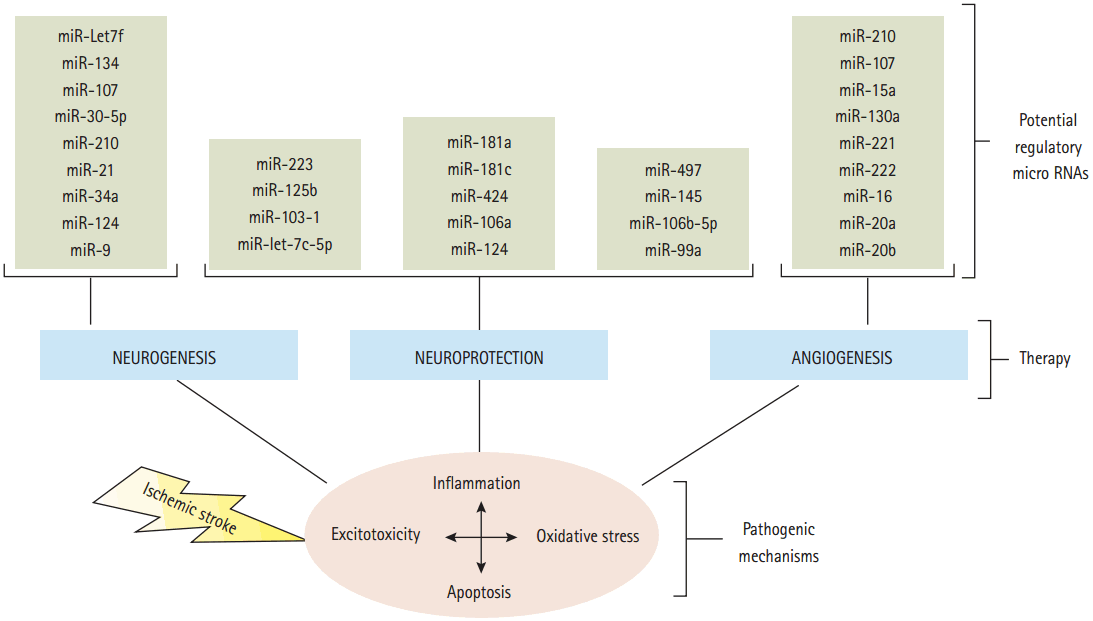
Overview of processes involved in ischemic stroke and high potential therapeutic microRNAs. Cerebral ischemia includes several injurious mechanisms (excitotoxicity, oxidative stress, inflammation and apoptosis) to confer neuronal injury. Potential therapeutic areas to compensate for these pathogenic process include promoting angiogenesis, neurogenesis and neuroprotective recovery and repair mechanisms.
MicroRNAs and neuroprotection
Neuroprotective strategies that limit secondary tissue loss and/or improve functional outcomes have been identified to help clinicians in decreasing stroke mortality rates and improving the quality of patient’s life [182]. Glutamate antagonists are the most studied neuroprotective agents. Glutamate is a major excitatory neurotransmitter in the CNS and is released excessively during ischemia [183]. miRNAs seem to offer some potential to attenuate excitotoxicity and miR-125b and miR-223 have been demonstrated to target NMDA receptor subunits including NR2A and NR2B, respectively, and negatively regulate their expression [53,54]. Hence, increasing the expression of this miRNAs represents a potential therapeutic application through decreasing the effects of excitotoxicity, which needs to be further investigated.
Calcium influx during ischemic stroke triggered intracellular destructive enzymes, which leads to brain tissue damage [184]. Interestingly, the sodium-calcium exchanger-1 (NCX1) gene expression is influenced by cerebral ischemia, which is a plasma membrane transporter that regulates cellular calcium and sodium homeostasis in the brain [185-187]. NCX activation ameliorates the consequences of ischemic brain damage [188]. So, it has been showed that anti-miR-103-1 exerts a strong neuroprotective effect against ischemic damage through NCX1 activation and offers the opportunity to develop a new therapeutic strategy for ischemic stroke [189]. Neuroprotection could also be achieved by targeting the inflammatory mediators that contribute to brain injury following ischemic stroke. miR-181a has deleterious effects on ischemic stroke, and using miR-181a antagomir caused neuroprotective effects, reduced NF-κB activation and improved neurological deficits in mice [190]. Furthermore, the ability to decrease brain ischemia injury (both focal and forebrain ischemia) makes miR-181a antagomir a therapeutic agent [163]. It has been shown that suppression of TLR4, which is mediated by miR-181c, could be neuroprotective in hypoxic injuries, so this offers a potential therapeutic agent for ischemic stroke associated with microglial activation [126]. Studies showed that miR-424 overexpression has a neuroprotective effect on cerebral ischemia injury through mechanisms relating to the preventing of microglia activation [116]. In the microglia and macrophages miR-106a and miR-124 leading to increasing in IL-10 and TGF-β respectively [118,134,135]. In turn, IL-10 and TGF-β inhibit expression of adhesion molecules in endothelial cells and production of pro-inflammatory cytokines [191,192]. Therefore, TGF-β and IL-10 are neuroprotective factors against neuro-inflammation [193-195].
As mentioned in the other sections, during ischemic stroke, there is increased production of reactive oxygen free radicals due to glutamate excitotoxicity [196]. Accordingly, the free radical scavengers potentially have neuroprotective roles. Preclinical studies in animal models that using those agents presented effectiveness in reducing neurological injuries [197]. miR-497 increased ischemic neuronal cell death with negatively regulating anti-apoptotic proteins, such as Bcl-2 and Bcl-w [152]. Antagonism of miR-497 leading to decreasing in the infarct volume due to ischemia in mice [152]. Some studies have shown that increasing Nrf2 activity is highly neuroprotective against ischemic consequences [74,198]. It has been shown that miR-145 antagomir increased protein levels of SOD2 after ischemic stroke [31]. It is determined that miR-106b-5p antagomir can protect against cerebral ischemia/reperfusion (I/R) injury by inhibition of apoptosis and oxidative stress [84]. Other studies indicate that miR-99a and miR-let-7c-5p have neuroprotective effects through inhibition of pro-caspase-3 and caspase-3 expressions as well as preventing apoptosis following cerebral ischemic stroke [158]. Sequestration of this miRNA could therefore serve as a potential defense against post-stroke pathogenic processes in neuroprotection therapy.
MicroRNAs and neurogenesis
Neurotrophic factors are small polypeptide molecules, which are involved in cell proliferation, migration, differentiation and development of the nervous system. In the adult CNS, neurotrophic factors have important roles in the survival and maintenance of neuronal cells by activating cell survival genes and inhibition of suicide genes [199]. For this reason, deprivation of these factors in the ischemic penumbra zone can trigger neuronal apoptosis and lead to cell death. In preclinical studies, neurotrophic factors such as nerve growth factor, brainderived neurotrophic factor (BDNF), ciliary neurotrophic factor, glialderived neurotrophic factor, vascular endothelial growth factor (VEGF), and insulin-like growth factor 1 (IGF-1) have all been shown to decrease infarct size in animal models [200]. Ischemic activated microglia can release a variety of cytoprotective substances by producing neurotrophic molecules such as IGF-1, BDNF, and several other growth factors [105,201].
It has been shown that miR-Let7f antagomir targets the IGF-1 signaling for translation activation which could alternatively promote IGF-1-like neuroprotection in the ischemic stroke models [202]. Downregulation of miR-134 alleviates ischemic injury through enhancing of BDNF and Bcl-2 expression in neurons following oxygen glucose deprivation. Therefore, miR-134 antagomir providing a potential therapeutic agent for cerebral ischemic injury [165]. miR-107 and miR-30-5p are reported as BDNF regulators and investigations into the precise mechanism of both these miRNAs in BDNF regulation can be a potential therapeutic agent in neuroprotection [203]. Overexpression of miR-210 can induce neurogenesis in the adult mouse brain, which is associated with VEGF upregulation [204]. VEGF is an important neurogenic factor with therapeutic potential in ischemic stroke [205].
Following cerebral ischemia, neuronal stem and precursor cells (NSC and NPC) can be activated and migrate to the injured areas [206]. Hedgehog, Notch, Wnt and TGF-β signaling pathways are found to be responsible for proliferation, migration and differentiation of NSC and NPC to promote neuronal repair after ischemic stroke [207-211]. miR-21 was found to be significantly upregulated following cerebral ischemia, and it could act as a NPC regulator by Wnt and TGF-β signaling pathways. Furthermore, miR-34a may negatively regulate the NPC proliferation by inhibiting Notch, Wnt, Hedgehog and TGF-β signaling pathways following brain ischemia [212]. It has been reported that increased miR-124 concentrations could promote neural differentiation by post-transcriptionally downregulation of Sry-Box 9 (Sox9). It is demonstrated that Sox9 overexpression abolished neuronal differentiation, whereas Sox9 knockdown led to improved neuron formation [213]. miR-124a was found to inhibit neurogenesis following stroke through targeting the JAG1/Notch signaling pathway. miR-124a in neural progenitor cells decreased JAG1 transcript and protein levels significantly, which causing to inactivation of Notch signals [214]. Furthermore, this microRNA was reported to be constitutively expressed in the brain mature neurons [215]. miR-9 has been revealed to limit migration and promote proliferation in human neuronal progenitor cells and its downregulation permits to neuronal migration [215]. Therefore, pharmacological regulation of these miRNAs could be a potential agent in the post-ischemic neurogenesis.
MicroRNAs and angiogenesis
Angiogenesis is an important, beneficial event occurring in ischemic stroke. Angiogenesis delivers blood flow and metabolism to ischemic tissue and is positively correlated with the survival rate of stroke patients [216]. miRNAs that regulate the process of angiogenesis have been offered as a potential treatment strategy for ischemic stroke [217]. Overexpression of miR-210 promotes focal angiogenesis in the adult mouse brain, which was associated with local increased VEGF levels [204]. Also, miR-210 can trigger vascular endothelial cell migration and tube formation under hypoxia in vitro [204]. Overexpression of miR-210 in patients with acute ischemic stroke show better clinical outcomes [218]. Hence, miR-210 is specifically sensitive to hypoxic stimuli in almost all of cells [219], and its expression is enhanced by hypoxia-related transcription factors, such as HIF-1α [220,221]. miR-92a regulates angiogenesis targeting several proangiogenic proteins, including the integrin subunit alpha5. Thus, miR-92a could be a therapeutic target in the setting of ischemic disease [222].
VEGF is an essential angiogenic factor with therapeutic potential in ischemic stroke [223,224]. It has been demonstrated that miR-107 contributes to post-stroke angiogenesis by directly down regulation of Dicer-1 expression which is a gene that encodes an important enzyme in the miRNA processing. This leads to translational de-suppression of VEGF mRNA, thereby increasing expression of VEGF (VEGF165/VEGF164), resulting in post-stroke angiogenesis [225]. miR-107 expression is regulated by HIF-1α and has binding sites with HIF-1α [226]. A novel finding indicates that overexpression of miR-15a in endothelial cells can suppress post-stroke angiogenesis via direct inhibition of endogenous endothelial fibroblast growth factor 2 and VEGF activities [227]. Also, studies have shown that expression of the miR-15a is significantly increased in the cerebral vasculature at the penumbral zone following cerebral ischemia [228]. Furthermore, miR-16, -20a and -20b have been found to target VEGF and act as an anti-angiogenic agent in cultured endothelial cells [229].
Growth arrest-specific homeobox (GAX) and homeobox A5 (HOXA5) are anti-angiogenic transcription factors and are involved in the inhibition of endothelial cell function. GAX is expressed in the endothelial cells and inhibits angiogenesis through down regulation of NF-kB signaling pathway [230,231]. miR-130a has been found to down-regulate GAX and HOXA5 expression, consequently antagonizing the antiangiogenic activity of these factors [232]. miR-221 and -222 were found to inhibit angiogenesis by interaction and down-regulation of kit ligand (KIT), and enriched of this microRNAs in the hippocampus of the mice indicates a possible role for them in stroke pathogenesis [233,234]. In the same way, it was suggested that miR-221 and miR-222 decreases tube formation and migration by targeting both KIT and endothelial NOS [235]. Therefore, pharmacological modulation of these miRNAs could be a promising therapeutic approach for angiogenesis after ischemic stroke.
Challenges for miRNA therapy
There is mounting evidence that miRNA-based therapies hold great promise. However, despite the exciting potential of miRNAs, critical hurdles remain to be overcome which often include delivery of miRNA-targeting agents. Other limitations including limited in vivo stability, limited tissue distribution, and untoward side effects. Although, either viral vectors and nonviral delivery systems such as liposomes could overcome these challenges, both liposomes and viral vectors may be toxic and/or immunogenic which would restrict their clinical application. Liposomes are utilized to deliver small interference RNAs (siRNA). However, synthetic systems such as liposomes have relatively lower yield compared to viral vectors [236,237].
After stroke, a high level of miRNAs leads to inhibition of the expressions of many genes. Therefore, inhibition of these miRNAs may be a therapeutic targets for ischemic stroke [238]. There are several tools to decrease the level of miRNA such as antagomir (anti-sense oligonucleotide), which blocks miRNA silencing activity by complimentary binding to the mature miRNA, and this could be a useful approach to inhibition of miRNA function [239]. Therefore, use of an antagomir may be another therapeutic option when upregulated miRNAs are pathogenic.
The advantage of antigomirs is that they can be delivered into cells directly without any vector assistant, because they are nuclease resistant. Therefore, antigomirs avoid the complication of using delivery vehicles. The drawbacks that limit antigomir application as therapeutic reagents in humans are the need for high doses and their possible side-effects [240-242].
Antagomirs could easily be delivered intravenously, but there is poor distribution in the brain due to the blood-brain barrier, which prevents most exogenous substances from entering the CNS [243,244]. In recent years, intranasal delivery has been used to target the brain, and evidence shows that olfactory nerve pathways, trigeminal nerve pathways, vascular and lymphatic pathways are involved in intranasal delivery [245]. Further studies have shown that intranasal delivery of antagomir- miR-206 reached the brain and increased memory function in mice with Alzheimer’s mice [246].
Furthermore, miRNAs have been introduced by mechanical methods such as high pressure injection and electroporation, but these methods cause too much damage to the tissues [247,248]. Administration of miRNAs in the absence of a carrier presents limited tissue distribution, and they are taken up by the liver and kidney and rapidly excreted in urine. In addition, the lethal dosage, LD50, of specific miRNAs has yet to be recognized [236]. Nevertheless, it is probable that an increasing number of these molecules will progress and will eventually be developed to become approved treatment for ischemic stroke in the coming years.
Conclusions
In this review we have presented evidence that miRNA function is increasingly dysregulated following ischemic stroke, and altering of these molecules has profound effects on the downstream target genes which are involved in the post-ischemic process. A single miRNA exerts its cellular function by mostly inhibition and occasionally activation of numerous downstream mRNA targets. Several studies have attempted to correlate between changes in the expression of miRNAs and postischemic pathogenic processes such as excitotoxicity, inflammation, oxidative stress and apoptosis. These studies clarify the contribution of miRNAs in the post-ischemic pathophysiological process and help us to a better understanding of the processes involved in ischemic stroke pathology, where they could be a therapeutic agent. Also, there is accumulating evidence that several miRNAs and their target genes are involved in the retrieval and repair process which including the promotion of angiogenesis, neurogenesis and neuroprotection.
miRNA profiles provide evidence that their modulation could be beneficial for ischemic stroke diagnosis, as well as being potential therapeutic agents. Moreover, the ability of miRNAs to regulate numerous target genes clearly demonstrates their importance in ischemic stroke therapeutics. Finally, the understanding of delivery systems will be a key to bringing miRNA to the clinic as findings from animal models become better refined to allow translation into human therapeutic agents for the treatment of ischemic stroke.
Notes
The authors have no financial conflicts of interest.
Acknowledgements
This work was supported by Ahvaz Jundishapur University Grant. The authors declare no financial or commercial conflict of interest.