Silent New Brain Lesions: Innocent Bystander or Guilty Party?
Article information
Abstract
With the advances in magnetic resonance imaging, previously unrecognized small brain lesions, which are mostly asymptomatic, have been increasingly detected. Diffusion-weighted imaging can identify small ischemic strokes, while gradient echo T2* imaging and susceptibility-weighted imaging can reveal tiny hemorrhagic strokes (microbleeds). In this article, we review silent brain lesions appearing soon after acute stroke events, including silent new ischemic lesions and microbleeds appearing 1) after acute ischemic stroke and 2) after acute intracerebral hemorrhage. Moreover, we briefly discuss the clinical implications of these silent new brain lesions.
Introduction
Although most strokes are considered a single event, subsequent strokes may follow shortly after an acute stressful event such as ischemic or hemorrhagic stroke. Remarkably, in many cases, such subsequent strokes after acute index events are asymptomatic. These silent new brain lesions can be demonstrated by diffusion-weighted imaging (DWI) and gradient echo T2* imaging (GRE) or susceptibility-weighted imaging. Although the name suggests clinically silent behaviors, their clinical importance remains controversial. While some researchers consider only clinically obvious stroke as clinically important [1], others have associated silent new brain lesions with future strokes or possible harmful effects on the brain function.
In this article, we aim to review silent new brain lesions in terms of their incidence, associated factors, mechanisms, and clinical implications. As both ischemic and hemorrhagic (microbleeds) strokes can appear as silent new brain lesions, this narrative review categorized silent new lesions into two groups: silent new ischemic lesions (SNILs) and silent new microbleeds (SNMs).
SNILs
SNILs after acute ischemic stroke
As mortality is increased in stroke patients with early recurrence [2,3], efforts have been made to identify patients at high risk of recurrence. To identify these patients in the early phase of the index stroke, DWI, which is the most sensitive imaging method for detecting hyperacute ischemic stroke [4,5], has been rigorously used to detect early recurrent ischemic lesions.
Definition and incidence
SNILs can be arbitrarily divided into early and late SNILs based on the time of magnetic resonance imaging (MRI). Early SNILs are usually defined as a new lesion on 5-day to 7-day DWI outside the region of the acutely symptomatic lesion (Table 1) [6-14]. Early SNILs are reportedly found in 24.2%-34.3% of patients with acute ischemic stroke [6-11], while higher rates have been reported in patients with large artery atherosclerosis [13,14] or in those receiving thrombolysis [12]. These rates of early SNILs largely exceed those of clinical evident recurrence, of which the cumulative rates have been reported to be 2% within 1 week [15] and 1.6%-8% in 90 days [16-18]. Accordingly, the majority of early SNILs are asymptomatic [6-10,13].

New ischemic lesions after acute ischemic stroke and minor stroke or TIA
Late SNILs occurring at a subacute stage have also been measured in several studies using DWI and fluid attenuated inversion recovery imaging at 30 or 90 days after the index stroke [7,8]. In these studies, the incidence of late SNILs was reported as 22.1%-26.3%, which is lower than that of early SNILs. However, the majority of late SNILs are also asymptomatic.
Characteristics
SNILs after acute ischemic stroke can be further categorized into two groups based on their locations, namely “local” or “distant” lesion recurrence according to whether a new lesion occurs within or outside the territory of initial perfusion deficit (Figures 1 and 2) [6,7]. In early SNILs, the proportion of local lesion recurrence is slightly higher than that of distant lesion recurrence (44.4%-51.9% vs. 44.1%-47.1%). On the contrary, in late SNILs, local lesion recurrence is less frequent than distant lesion recurrence (23.8% vs. 76.2%) [7].
Early local silent new ischemic lesions (SNILs). Acute diffusion-weighted imaging (DWI) was performed within 24 hours after symptom onset and follow-up DWI was performed 2 days after the index ischemic stroke. Early local SNILs (arrows) are shown on the follow-up DWI.

Early distant silent new ischemic lesions (SNILs). Acute diffusion-weighted imaging (DWI) and perfusion-weighted imaging were performed within 24 hours after symptom onset. Follow-up DWI was performed 8 days after the index ischemic stroke. Early distant SNILs (arrows) are indicated on the follow-up DWI.
Moreover, the lesion size of most SNILs is small [12,13], with the reported lesion volumes ranging between 0.3 and 0.7 mL [13], and the majority of SNILs (70.6%) being 10 mm or less in diameter [12]. These findings are consistent with the fact that most recurrent ischemic lesions do not cause any evident symptoms in the patients.
Associated factors and mechanism
Multiple ischemic lesions on baseline DWI are significantly associated with early SNILs [6,9,10]. If multiple acute lesions are demonstrated on clinical presentation, especially with varying degree of apparent diffusion coefficient (e.g., reduced and normalized), those lesions may have occurred at varying time points before or after the clinical stroke onset [6,19]. Moreover, increased microembolic events have been reported to be related with these multiple lesion patterns [20].
Among the different stroke subtypes, large-artery atherosclerosis (LAA) has been most frequently associated with early SNILs [6,8-10,13], and has been shown as an independent predictor of SNILs after acute ischemic stroke [10]. In particular, early SNILs in intracranial LAA have different characteristics compared to other stroke subtypes [13]. Early SNILs in intracranial LAA occur mostly in the pial area of the same vascular territory as the index stroke and are more frequently observed in higher grades of stenosis. On the other hand, in extracranial LAA, the degree of stenosis is not related to early SNILs, and these are not associated with subsequent recanalization, whereas in cases of cardioembolism, early SNILs are associated with significant recanalization. In intracranial LAA, artery-to-artery embolism or hemodynamic insufficiency may play an important role in the pathogenesis of recurrent ischemic stroke [21]. Meanwhile, plaque heterogeneity and fragmentation of the initial embolus may be more crucial in the pathogenesis of SNILs in extracranial LAA and cardioembolism, respectively [22]. Lastly, early SNILs in intracranial LAA are more closely associated with clinical recurrence than in the other subtypes. In line with these findings, patients with stroke resulting from intracranial LAA have been demonstrated to show a high risk of recurrent stroke (>20% over 2 years) [23].
Thrombolytic therapy and vessel recanalization have also been shown to be associated with early SNILs [9,12]. Chronologically, recombinant tissue-type plasminogen activator treatment is associated with acute SNILs, which occur between 24 and 48 hours, while spontaneous vessel recanalization is associated with subacute SNILs, occurring between 2 and 6 days [9]. Recombinant tissue-type plasminogen activator treatment can contribute to the occurrence of SNILs by imperfect dissolution of either the embolus at the site of the main vessel occlusion or the thrombus at the site of origin; however, its short half-life may limit its role to only the acute phase.
Regarding the baseline hypoperfusion status, large mild perfusion delays have been shown to be independently associated with early SNILs. Meanwhile, more severe perfusion delays, together with large initial DWI lesion, have been demonstrated to be related with infarct lesion growth in the subsequent DWI within 7 days [12].
Among the various plasma or serum biomarkers measured within 24 hours after the onset of index stroke, an elevated level of D-dimer has been shown to be independently associated with early SNILs at 5-day [10]. Increased D-dimer levels may reflect ongoing thrombus formation within cerebral vessels or systemic hypercoagulability [24]. Moreover, D-dimer itself may induce the inflammatory process by stimulating monocyte synthesis and release of proinflammatory cytokines such as interleukin-6 [25].
Insufficient inhibition of platelet aggregation by the administered drugs may also contribute to the advent of SNILs [11,14]. Biochemical aspirin resistance, defined as an aspirin reaction unit ≥550 (VerifyNow Aspirin Assay), was associated with distant early SNILs occurring outside the vascular territories of index stroke in one previous study [11]. In addition, a certain genotype of cytochrome P450 2C19, which poorly metabolizes clopidogrel into its active form, has been reported to relate with a significantly higher rate of SNILs in patients with stroke due to LAA [14].
Taken together, the mechanisms of early or late SNILs are diverse (Figure 3), and they can be categorized according to the locational distribution of SNILs. For local lesion recurrence, several mechanisms are plausible, including 1) fragmentation of the initial embolus during the process of recanalization and reperfusion [6]; 2) recurrent ischemic events (recurrent emboli) within the same perfusion deficit caused by intrinsic atherosclerosis [6]; and 3) recurrent ischemic events due to prolonged hypoperfusion [11]. Meanwhile, for distant lesion recurrence, recurrent embolic events arising from a proximal source, such as the heart or activated atherosclerotic plaque located outside of the initial culprit vessels, may be important. Accordingly, atrial fibrillation and the presence of microembolic signals demonstrated by transcranial Doppler ultrasonography are reportedly associated with the advent of early SNILs [20]. Of note, there are also other players acting as common pathogenic denominators. For example, induced inflammatory response after stroke and/or hypercoagulability, which sustainably activate plaque, may contribute to the development of both local and distant SNILs [6].
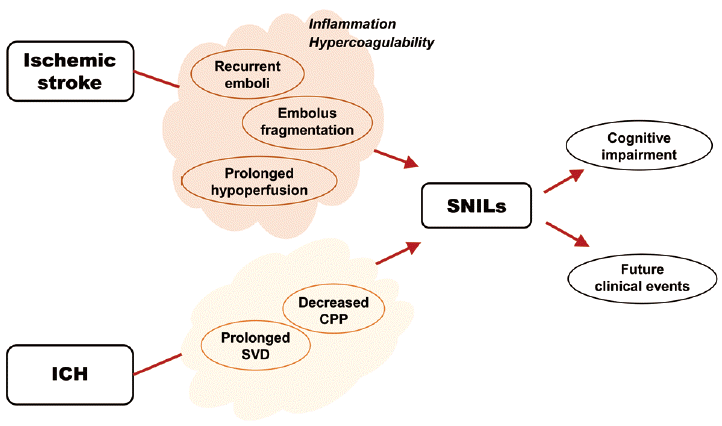
Pathophysiology and clinical implications of silent new ischemic lesions (SNILs) after stroke. CPP, cerebral perfusion pressure; ICH, intracerebral hemorrhage; SVD, small vessel disease.
Early and late SNILs and clinical vascular events
Early SNILs at 5 days have been shown to be independently associated with late SNILs at 30 or 90 days, suggesting a continued risk of recurrent ischemic lesions in the weeks following the index stroke [7]. Atheroma, which is an acute-on-chronic disease causing recurrent episodes of thromboembolism before the stabilization of an ulcerated plaque, has been suggested as the culprit mechanism for this prolonged stroke-prone state [26].
As for clinical vascular events, late SNILs independently predict recurrent ischemic strokes. Early SNILs, together with late SNILs, can predict the combined clinical endpoints of recurrent ischemic stroke, transient ischemic attack (TIA), and vascular death [8]. The superiority of late SNILs to early SNILs in predicting subsequent clinical vascular events may be explained by the different pathogeneses of the two types of SNILs. Early SNILs, which include local lesion recurrence, may result from progression of the initial ischemic event. On the contrary, late SNILs, of which the major form is distant lesion recurrence, may accurately reflect the risk of future recurrent ischemic events [8].
Minor stroke and TIA
It has been considered that a previous TIA or minor stroke confers a greater risk for recurrent stroke [27,28]. Accordingly, SNILs can also appear after TIA and minor stroke (National Institutes of Health Stroke Scale score <3-6) [29-31]. The incidences of SNILs in this population have been reported as 18% at 7 days, and 7%-22% at 30 or 90 days, both of which are lower than those of SNILs after acute ischemic stroke (Table 1). This difference may be attributable to the fact that TIA and minor strokes are less likely to recur compared to acute ischemic strokes [29].
Further, LAA and cardioembolism are most likely to be associated with SNILs in patients with TIA and minor strokes [19,29]. Among the MRI parameters, the baseline DWI lesion volume and perfusion deficits have been shown to significantly associate with the advent of SNILs [30,31].
SNILs after acute intracerebral hemorrhage (ICH)
Since ischemic and hemorrhagic strokes are distinctive diseases, stroke patients usually present with one of the two. However, although rare, symptomatic acute ischemic and hemorrhagic stroke can occur simultaneously or shortly after one another [32-34]. Moreover, both types of stroke share common risk factors such as old age, hypertension, and high alcohol intake [35,36]. In this regard, there have been efforts to investigate SNILs after acute ICH using DWI.
Definition, incidence, and baseline characteristics
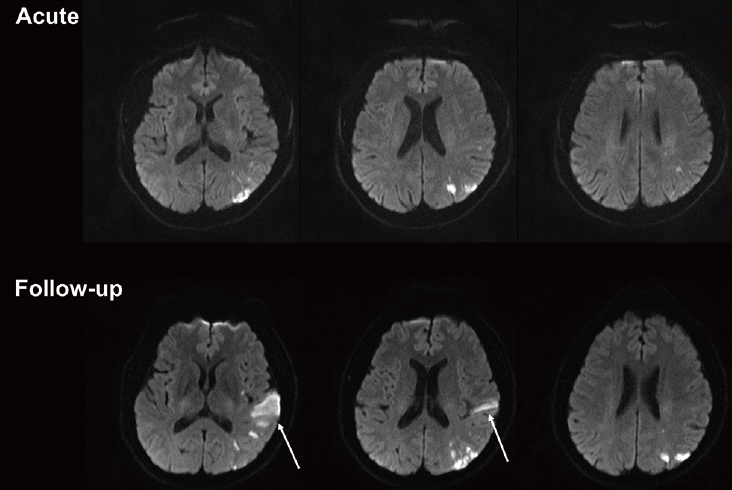
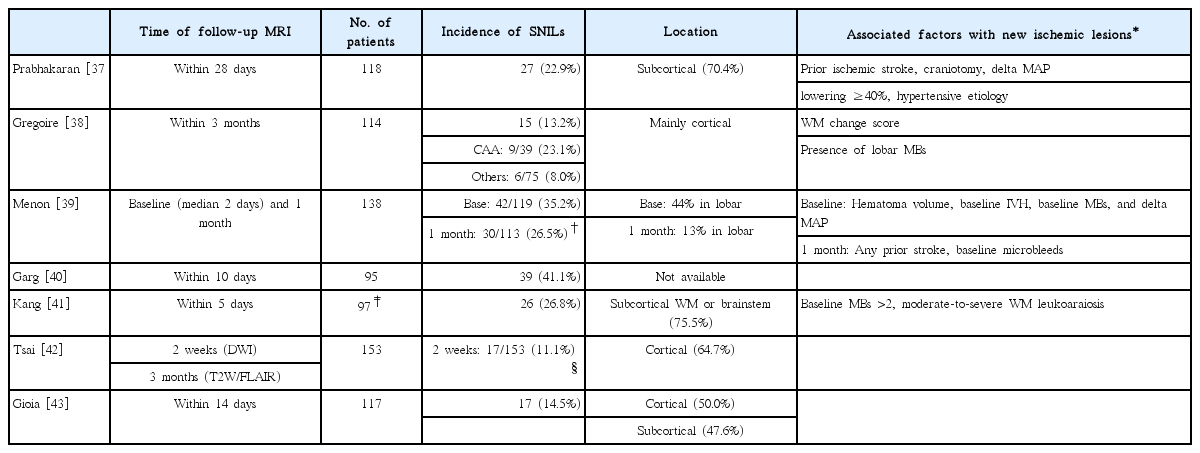
SNILs after acute ICH are defined as any hyperintense lesions on the subsequent DWI that are distinct from hemorrhage and the area of perihematomal edema (Figure 4) [37-43]. SNILs may appear as early as within 5 days; follow-up MRI is usually performed within 14 days, but may be performed up to 3 months after the index hemorrhage (Table 2). The reported prevalence rates of SNILs after acute ICH are 11.1%-41.1% within 14-days, 22.9%-26.5% at 1 month, and 13.2% at 3 months. The lesion size of SNILs after ICH also tends to be small; more than 70% of lesions have a diameter less than 10 mm [37,38,42]. Further, patients with cerebral amyloid angiopathy are more likely to develop SNILs at 3 months after ICH, and have a more severe hemorrhagic burden (i.e., more frequent microbleeds, multiple ICHs, and intraventricular hemorrhage) compared to those with other primary ICHs [38]. The main locations of SNILs have been reported to vary. Notably, however, patients with hypertensive ICH develop SNILs mainly in the subcortical white matter or brainstem [41].

Early silent new ischemic lesions (arrows) coexisting with acute intracerebral hemorrhage in the left basal ganglion. Magnetic resonance imaging was performed 2 days after symptom onset.
New ischemic lesions after acute intracerebral hemorrhage
The natural course of SNILs after acute ICH is dynamic. When comparing baseline (median, 2 days) and 1-month DWIs after acute primary ICH, more than 80% of SNILs on the follow-up DWI were not present at baseline, demonstrating that SNILs develop through an ongoing process during the acute phase [39]. Meanwhile, half of all SNILs on the 2-week DWI reportedly do not remain on the subsequent 3-month MRI, suggesting that not all early SNILs after acute ICH indicate permanent tissue injury [42].
Associated factors and mechanisms
Several risk and clinical factors have been reported to be assoassociated with SNILs after acute ICH, although the results are inconsistent and even contradictory among studies.
Microbleeds and leukoaraiosis have been independently and consistently associated with the occurrence of SNILs after acute ICH [38,39,41]. Microbleeds are an imaging marker of bleeding-prone microangiopathies [44], while leukoaraiosis represents chronic hypoperfusion in the distal deep and arteriolar territories [45]. Remarkably, microbleeds and leukoaraiosis are strongly correlated [46], and, when combined, they represent small vessel pathology [47]. Chronic changes by longstanding hypertension in the form of microaneurysm, lipohyalinosis, and fibrinoid degeneration develop in the deep small vessels and are associated with both hypertensive ICH and ischemic lacunes. Thus, an active small-vessel angiopathic process induced by hemorrhagic insults may underlie the advent of SNILs [41].
History of ischemic stroke has been suggested as an independent predictor of SNILs after acute ICH [37,39]. Moreover, this has been regarded as a surrogate marker of perforator disease, where relative hypoperfusion can result in ischemia in a single or multiple perforators [37]. However, the association between prior ischemic stroke and SNILs after acute ICH was denied in other studies [40,41].
Another suggested risk factor of SNILs after acute ICH is decreased blood pressure during the acute phase [37,39,40]. In chronic hypertension, the curve of cerebral autoregulation is shifted to the right, and after acute brain injury, autoregulation may be abolished so that cerebral blood flow changes linearly according to cerebral perfusion pressure [48]. As elevations in intracranial pressure due to acute ICH would result in cerebral perfusion pressure decreases, aggressive blood pressure lowering therapy beyond the lower limits of cerebral autoregulation might induce cerebral ischemia in ICH patients with chronic hypertension [37]. However, of note, the perihematoma region is known to be unaccompanied by hypoperfusion or ischemia, and rather shows benign oligemia [49-53]. Moreover, the relationship between reduction of blood pressure and the advent of SNILs was not reproduced in some studies [41,43]. Thus, it remains controversial whether decreased blood pressure is the culprit behind the occurrence of SNILs.
Large ICH volume and subsequent craniotomy have been suggested to be associated with SNIL development [37,39]. Elevated intracranial pressure in patients with large volume of ICH may have a negative impact on the cerebral perfusion pressure and secondarily on the cerebral blood flow. In addition, brain shifts from large hematomas may directly compress neighboring vascular structures, contributing to tissue infarction. However, both large ICH volume and craniotomy did not show any significant association with SNILs in other studies [40,41].
Clinical implication of SNILs
Predictor of prognosis and future events
SNILs on DWI after acute ischemic or hemorrhagic stroke can be used to predict future clinical events and prognosis. DWI can be considered as a crystal ball, while SNILs are an omen.
Regarding SNILs after acute ischemic stroke, both early SNILs at 5 days and late SNILs in the subacute phase up to 90 days have been shown to be independent predictors of subsequent clinical vascular events such as recurrent ischemic stroke, TIA, and vascular death during a median follow-up of 1.6 years [7,8]. Thus, patients with SNILs over the early weeks after an index stroke should be considered optimal candidates for early aggressive stroke prevention therapy. In addition, SNILs may provide a useful surrogate endpoint in clinical trials evaluating stroke prevention therapies, with the reduction in the lesion recurrence rate upon pharmacologic intervention over the initial weeks used as a surrogate for reduction in clinical stroke recurrence over the following years. Clinical trials to test the efficacy of therapeutic interventions usually take many years to recruit for and complete. An MRI surrogate endpoint of recurrent stroke, which would allow substantially fewer patients and shorter follow-up period, would result in enormous savings of cost and time in evaluating the preventive therapies. However, currently, only data from single-center and retrospective studies are available. Thus, future prospective randomized controlled trials testing the effect of a stroke prevention therapy on SNILs and clinical stroke recurrence in the long-term are required.
SNILs after acute ICH also have prognostic value; in one study, they were independently associated with dependence or death of patients at 3 months [40], and in another, with the comcomposite of clinical cerebrovascular endpoints (i.e., ischemic stroke, ICH, and vascular death) during a median follow-up of 3.5 years [41]. These findings warrant special care in ICH patients with SNILs to reduce the risk of cerebrovascular events in the future. As SNILs after acute ICH predict not only hemorrhagic but also ischemic strokes during the follow-up, antiplatelets associated with lower risks of bleeding complications may be considered in this group of patients to prevent subsequent cerebrovascular events. The efficacy of antiplatelets in these patients should be proven, and the safe interval from ICH onset to initiation of antiplatelet administration is also to be determined. In addition, the optimal level of blood pressure should be delicately set to prevent SNILs as well as ICH progression.
Cognitive impairment
Silent ischemic lesions on MRI have been associated with cognitive dysfunction or dementia in asymptomatic populations [54-67]. The size and location of ischemic lesions may be important for such associations. In order to remain silent, the ischemic lesions should be small and located in appropriate areas such as the frontal lobe, not causing focal neurologic deficits. However, as silent lesions accumulate in the affected area, the increased ischemic burden may contribute to impairment of cognitive function and to difficulties in mental flexibility, language, and memories [68-74]. Besides the location, the extent of silent ischemic lesions is also important for the development of cognitive dysfunction. The degree of the lesions on MRI has shown a positive correlation with the severity of cognitive impairment [65,75-77].
SNILs after acute ischemic or hemorrhagic strokes are small and asymptomatic initially; further, they can occur virtually anywhere according to the location of the index stroke. Thus, their accumulation may also contribute to cognitive decline during the follow-up. Moreover, considering that many patients with a history of ischemic or hemorrhagic strokes are also accompanied by many vascular risk factors, which are known to increase the risk of dementia [78], special attention should be paid to the risk of cognitive impairment in these patients.
SNMs
Microbleeds refer to small, round, dark-signal lesions detected by T2*-weighted GRE or susceptibility-weighted imaging [79-81]. They are known to be manifestations of focal extravascular leakage of blood components, representing old microhemorrhages, but rarely cause overt symptoms [82]. Although not studied as much as SNILs, SNMs are also known to appear after acute ischemic or hemorrhagic stroke.
SNMs after acute ischemic stroke
Microbleeds are frequently found in ischemic stroke patients, at varying rates (35%-71%) [82-84]. In addition, they are more common in patients with recurrent strokes than in those experiencing a first-ever stroke [85], whereas they are rarely found in TIA patients [86]. These findings suggest that ischemic stroke may trigger the occurrence of microbleeds.
Definition and incidence
Microbleeds are defined as unambiguous homogeneous, round, signal-loss lesions with diameters up to 5-10 mm and with blooming artifacts, as determined by GRE [87-89]. In one previous study, within 24 hours, approximately 30% of patients with acute ischemic stroke showed a number of concurrent microbleeds (median 2, range 1-33) in their brains [87]. During the acute phase, another 13% of patients developed SNMs (median 1, range 1-5) on the 7-day GRE, while 3% of patients lost their baseline microbleeds [87]. As for the locations, about half of all microbleeds were detected in the deep hemisphere; at baseline, 32% were located in the lobar location while 30% were in the deep location, whereas on follow-up GRE, 52% of SNMs were found in the lobar location [87].
The long-term fate of SNMs is also dynamic, as the appearance and disappearance of SNMs can occur years after the index acute ischemic stroke [88,89]. In one study, during a mean follow-up of 27 months, 38% of acute ischemic stroke patients had SNMs on their subsequent GRE, with the total number of microbleeds increasing by about 1.5-fold [88]. Among the patients with baseline microbleeds, 54% developed SNMs during their follow-up, showing a generally increasing trend in the number of microbleeds, although some of these lesions disappeared in 15% of those patients. In fact, de novo SNMs were detected even on the 5-year follow-up GRE in 30% of patients with index ischemic strokes [89].
Associated factors and mechanism
The presence and number of baseline microbleeds are consistently associated with the advent of SNMs on follow-up GREs, regardless of the time from baseline (5 days to 5 years) [87-89]. Moreover, they have been shown to be positively related with the annual rate at which SNMs develop [88]. The multiplicity of microbleeds at the initial clinical presentation may imply that they occurred through various time points around the onset of stroke, suggesting a prolonged risk of SNMs in particular stroke patients [87]. The number of baseline hemorrhages may be a marker of the severity and aggressiveness of the underlying vascular disease [87]. In addition, the above findings are in line with findings of other studies indicating that the total number of baseline microbleeds can predict the risk of future hemorrhages [90-92]. The burden of small vessel disease such as leukoaraiosis or lacunar infarctions is an independent predictor of the development of SNMs in ischemic stroke patients [87,88]. The small vessel disease severity is well known to correlate with the number of microbleeds [46]. As microbleeds arise from microangiopathic changes after chronic hypertension, the number of microbleeds may hence reflect bleeding-prone and small vessel disease-prone microangiopathy [93].
The level of low-density lipoprotein cholesterol is inversely associated with SNMs [88]. Low concentrations of serum cholesterol are related with the risk of microbleeds, suggesting an antagonizing role of cholesterol in the pathogenesis of microbleeds [94,95]. Given that microbleeds is characterized by hemosiderin pigment accumulations in macrophages adjacent to the ruptured atherosclerotic microvessels, the low-density lipoprotein cholesterol level may be related to either changes of microaneurysms [94,96] or the clearance of hemosiderin-containing microglia [88].
Increased body temperature has also been reported to be associated with the presence of SNMs within 7 days [87]. High temperature is known to induce blood-brain barrier disruption after ischemic stroke [97]. As endothelial activation and damage, with subsequent breakdown of the blood-brain barrier, are key features in cerebral small vessel diseases, high temperature may also contribute to the disease process [98].
However, a caution in interpretation is needed for the above-mentioned studies, since spatial registration of baseline and follow-up GRE images have not been performed in any published studies [82].
SNMs after acute ICH
The coexistence of SNMs in patients with ICH has consistently been reported as more frequent than that in ischemic stroke patients, reaching 50%-80% [91,93,99-101]. However, only limited studies have evaluated whether SNMs occur following acute ICH by performing serial GRE studies.
In one study, SNMs occurred in 9 of 24 patients (38%) with cerebral amyloid angiopathy and previous lobar hemorrhage during the 1.5-year follow-up period [102]. In another study, in hypertensive ICH patients, SNMs were found in 19 of 63 patients (30%) after a median follow-up of 23 months [90].
Clinical implication of SNMs
Since its discovery, the clinical significance of microbleeds has been actively investigated. The presence of microbleeds is associated with the future advent of hemorrhagic stroke after index ischemic or hemorrhagic stroke, especially in patients with severe white matter ischemic lesions [90,103,104]. In addition, microbleeds have been associated with larger volume of ICH [105,106] and hemorrhagic transformation after ischemic stroke [107-109], although the latter has been denied by other studies [110-114]. Recurrent ischemic stroke also has been linked to the presence of microbleeds [115], while the association turned out to be only modest when compared to that of hemorrhagic stroke in a systematic review [103]. Lastly, microbleeds are associated with cognitive dysfunction, especially frontal-executive impairment [116,117].
Based on the above-mentioned data, SNMs appearing after acute cerebrovascular events may be suggestive of future risk of hemorrhagic stroke and cognitive dysfunction, similar to SNILs. However, there is currently no direct evidence that SNMs can predict these events in the early phase after acute events. Thus, SNMs should be examined in more detail in terms of their clinical implications, and future studies are warranted.
Notes
This research was supported by the National Research Foundation of Korea Grant NRF-2014R1A2A1A11051280 funded by the Korea government, and the Korea Health Technology R&D Project, Ministry for Health & Welfare, Republic of Korea Grants HI12C1847 and HI14C1983.
Notes
The authors have no financial conflicts of interest.